








Abstract – Unkritische Medizinprodukte: MDR, Klasse I & Betreiberpflichten
- Trotz ihres risikoarmen Status unterliegen Klasse-I-Medizinprodukte seit der MDR (EU 2017/745) und der MPBetreibV-Novelle 2025 verschärften Anforderungen; Praxisinhaber haften persönlich nach §§ 11, 92 MPDG bei Verstößen.
- Die Subklassen Is (steril), Im (Messfunktion) und Ir (wiederverwendbare chirurgische Instrumente) erfordern abweichend vom Grundsatz der Selbstzertifizierung eine externe Prüfung durch eine Benannte Stelle, erkennbar an der vierstelligen Kennnummer hinter dem CE-Zeichen.
- Die MPBetreibV 2025 verpflichtet Betreiber zur Einweisung bei jedem Produkt mit Gebrauchsanweisung (auch Klasse I), zur regelmäßigen Prüfung von IT-Sicherheitsaspekten vernetzter Produkte sowie zur unverzüglichen Meldung von Vorkommnissen an das BfArM.
- Ab dem 28. Mai 2026 ist die Registrierung neuer Produkte in EUDAMED verpflichtend; Betreiber können dort via UDI-DI die Konformität digital verifizieren und so Beschaffungsrisiken sowie Haftungsexposition minimieren.
Inhaltsverzeichnis
Was sind unkritische Medizinprodukte — Definition nach MDR
Der Begriff „unkritisch“ stammt primär aus der hygienischen Klassifizierung der KRINKO am Robert Koch-Institut (RKI). Es beschreibt Produkte, die lediglich mit intakter Haut in Berührung kommen. Im regulatorischen Sinne der MDR EU 2017/745 entsprechen diese meist der Klasse I — der Stufe mit dem niedrigsten Risikopotenzial.
All non-invasive devices are classified as class I, unless one of the rules set out hereinafter applies.
Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC (Text with EEA relevance. )
OJ L 117, 5.5.2017, p. 141.
Die MDR definiert Klasse-I-Produkte über eine Ausschlusslogik: Ein Produkt fällt in Klasse I, wenn keine der Regeln 2–22 nach (Anhang VIII) greift, die eine höhere Einstufung erzwingen würden. Typisch sind drei Merkmale: kein invasiver Kontakt, keine aktive Energieabgabe und kein lebenserhaltender Zweck.
Abgrenzung der Risikoklassen
Die Spaulding-Klassifizierung orientieren sich an Invasivität, Kontaktdauer und dem Patientenrisiko:
| Klasse (MDR) | KRINKO-Kategorie | Risikoniveau | Beispiele |
| I | Unkritisch | Gering | Rollstühle, Gehhilfen, Stethoskope |
| IIa | Semikritisch | Mittel | Hörgeräte, Ultraschallgeräte |
| IIb | Semikritisch bis Kritisch | Erhöht | Beatmungszubehör, Defibrillator-Paddles, Endoskope |
| III | Kritisch | Hoch | Koronarstents, Herzschrittmacher, chirurgisches Besteck |
Hinweis: Während bei Klasse IIa und höher eine Benannte Stelle (Notified Body) zwingend ist, genügt bei der Standard-Klasse I die Selbstzertifizierung des Herstellers — mit Ausnahme der Subklassen Is, Im und Ir.
Hier erfahren Sie mehr über die Risikobewertung von Medizinprodukten.
Klassifizierungsregeln für unkritische Medizinprodukte nach MDR
Anhang VIII MDR enthält 22 Klassifizierungsregeln. Für Klasse-I-Produkte sind die Regeln 1 bis 4 entscheidend, da sie nicht-invasive Produkte und chirurgisch-invasive Produkte zur vorübergehenden Verwendung adressieren.
Die 4 Regeln im Überblick:
- Regel 1: Erfasst alle nicht-invasiven Produkte.
- Regel 2: Bezieht sich auf die Ableitung/Lagerung von Körperflüssigkeiten.
- Regel 3: Alle nicht-invasiven Produkte zur Änderung der biologischen oder chemischen Zusammensetzung von Blut oder Geweben.
- Regel 4: Bezieht sich auf alle nicht-invasiven Produkte, die mit verletzter Haut oder Schleimhaut in Berührung kommen.
Wichtig: Der Verwendungszweck bestimmt die Klasse. Ein Handschuh kann als Medizinprodukt (MDR) und gleichzeitig als Persönliche Schutzausrüstung (PSA-Verordnung) gelten (Dual-Use).
| Regel (Anhang VIII) | Beschreibung | Klassifizierung | Praxisbeispiele |
| Regel 1 | Alle nicht-invasiven Produkte, sofern keine anderen Regeln greifen | Klasse I | Rollstühle, Gehhilfen, Krankenhausbetten |
| Regel 2 | Nicht-invasive Produkte zur Ableitung oder Lagerung von Körperflüssigkeiten oder Geweben | Klasse I (mit Ausnahmen) | Urinsammelbeutel, Probenbehälter |
| Regel 3 | Nicht-invasive Produkte zur Änderung der biologischen oder chemischen Zusammensetzung von Blut oder Geweben | Klasse IIa / IIb | Hämodialyse-Geräte, Blutbeutel |
| Regel 4 | Nicht-invasive Produkte, die mit verletzter Haut oder Schleimhaut in Berührung kommen | Klasse I / IIa / IIb | Pflaster, Wundauflagen (je nach Tiefe der Wunde) |
Die Subklassen der Klasse I im Überblick
Die MDR differenziert innerhalb der Klasse I drei Sondersubklassen, bei denen entgegen dem allgemeinen Grundsatz der Selbstzertifizierung eine externe Prüfung erforderlich ist:
- Klasse Is (Steril): Produkte, die in sterilem Zustand in den Verkehr gebracht werden (z. B. sterile Verbände). Die Benannte Stelle prüft hierbei die Herstellung und Aufrechterhaltung der Sterilität.
- Klasse Im (Messfunktion): Produkte mit einer Messfunktion (z. B. Fieberthermometer oder Messbecher). Hier konzentriert sich die Prüfung auf die messtechnischen Anforderungen und die Genauigkeit.
- Klasse Ir (Wiederverwendbare chirurgische Instrumente): Instrumente wie Pinzetten oder Scheren bleiben Klasse I, erfordern aber eine Prüfung der Aufbereitungsanweisung durch eine Benannte Stelle.
Hinweis: Für den Betreiber (Arztpraxis oder Krankenhaus) bedeutet dies, dass bei diesen Produkten zwingend eine vierstellige Kennnummer der Benannten Stelle hinter dem CE-Kennzeichen auf der Verpackung oder dem Produkt angebracht sein muss. Wer Ir-Produkte ohne die vierstellige Kennnummer der Benannten Stelle hinter dem CE-Zeichen nutzt, handelt ordnungswidrig nach § 94 MPDG.
Konkrete Beispiele unkritischer Medizinprodukte in der Arztpraxis
| Produktkategorie | Beispiel | Subklasse | Besonderheit |
| Instrumente | Pinzette, Skalpellgriff | Ir | Benannte Stelle für Instrumentenaufbereitung nötig |
| Verbandmaterial | Mullbinden, Pflaster | I | Nicht-steril: Selbstzertifizierung |
| Schutzausrüstung | Einmalhandschuhe | I | Parallel PSA-Verordnung prüfen |
| Diagnostik | Stethoskop, Reflexhammer | I | RKI-Einstufung: “Unkritisch” |
| Apparate | Digitales Blutdruckgerät | I | Achtung: IT-Sicherheitspflichten |
Grenzfälle: Wann wechselt ein Produkt die Klasse?
Der Verwendungszweck bestimmt die Klassifizierung — nicht das Produkt an sich. Ein Handschuh, der als Schutzausrüstung vertrieben wird, unterliegt der PSA-Verordnung. Wird er als Medizinprodukt mit medizinischem Verwendungszweck vertrieben, greift die MDR. Auch ästhetische Produkte ohne medizinischen Zweck (z. B. Filler, Anhang XVI MDR) müssen wie Medizinprodukte behandelt werden.
Tipp: Prüfen Sie bei kombinierten Produkten — z.B. Wundauflagen mit Wirkstoffzusatz — immer den in der Konformitätserklärung angegebenen Verwendungszweck. Weicht Ihr tatsächlicher Einsatz davon ab, erlischt der Haftungsschutz des Herstellers.
Pflichten für Hersteller und Betreiber nach MDR
Rolle des Herstellers & EUDAMED
Der Hersteller erstellt die Konformitätserklärung. Ab dem 28. Mai 2026 ist die Registrierung neuer Produkte in der EUDAMED-Datenbank verpflichtend. Betreiber können dort via UDI-DI die Konformität prüfen.
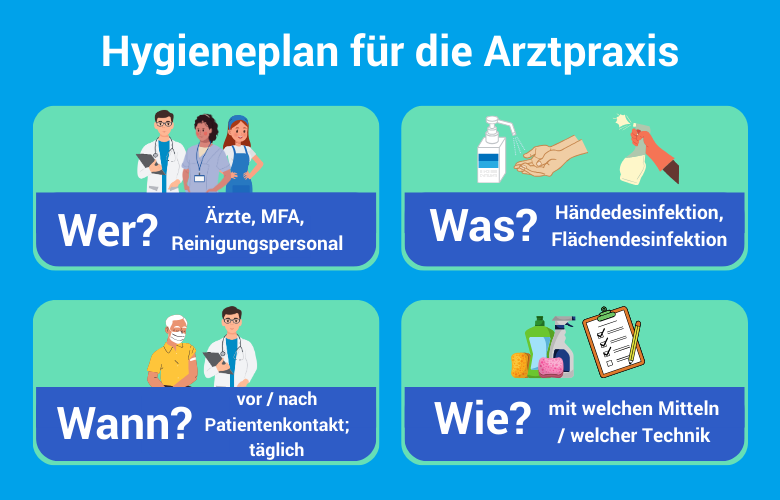
Pflichten des Betreibers (Praxisinhaber): Als Betreiber unterliegen Sie dem MPDG und der MPBetreibV:
- Einweisungspflicht (§ 4 MPBetreibV): Die MPBetreibV schreibt vor, dass Personen, die Medizinprodukte benutzen, in deren sachgerechte Handhabung eingewiesen sein müssen. Die Ausnahme für „selbsterklärende“ Produkte wurde verschärft; liegt eine Gebrauchsanweisung vor, ist die Einweisung i. d. R. Pflicht.
- IT-Sicherheit (§ 17 MPBetreibV): Mit der zunehmenden Vernetzung von Medizinprodukten müssen Betreiber nun sicherstellen, dass Software-Updates und IT-Sicherheitsaspekte regelmäßig geprüft werden. Dies ist eine direkte Reaktion auf die steigende Zahl von Cyberangriffen auf Gesundheitseinrichtungen.
- Bestandsverzeichnis (§ 14): Die Verpflichtung zur Führung eines Bestandsverzeichnisses für alle aktiven nicht-implantierbaren Medizinprodukte bleibt bestehen, wobei die Anforderungen an die einzutragenden Daten gestrafft wurden, um den bürokratischen Aufwand zu reduzieren.
- Meldepflicht: Vorkommnisse sind unverzüglich dem BfArM zu melden.
Die strafrechtliche Verantwortung des Betreibers nach MPDG
Das MPDG enthält in den §§ 92 bis 94 drastische Straf- und Bußgeldvorschriften, die den Schutz von Patienten und Benutzern sicherstellen sollen.
- § 92 MPDG (Strafvorschriften): Wer entgegen § 11 MPDG ein Medizinprodukt betreibt oder benutzt, das Mängel aufweist, durch die Patienten gefährdet werden können, riskiert eine Freiheitsstrafe von bis zu drei Jahren oder eine Geldstrafe.
- § 94 MPDG (Bußgeldvorschriften): Ordnungswidrigkeiten können mit Geldbußen bis zu 30.000 Euro geahndet werden (§ 94 Abs. 5). Dazu gehört beispielsweise:
- Das Fehlen einer ergänzenden Anzeige (§ 4 MPDG).
- Die Nichtbeachtung von Einweisungspflichten (jetzt explizit auch nach relevanten Software-Updates) oder IT-Sicherheitsanforderungen der MPBetreibV 2025.
- Der Betrieb von Produkten nach Ablauf des Datums, bis zu dem eine sichere Verwendung möglich ist (§ 12 MPDG)
Unkritische Medizinprodukte beschaffen: Was Arztpraxen beachten müssen
Checkliste: 6 Punkte vor der Beschaffung
- CE-Kennzeichen auf dem Produkt oder der Verpackung sichtbar
- EU-Konformitätserklärung beim Lieferanten verfügbar (bei Is/Im: benannte Stelle angegeben)
- Verwendungszweck in der Produktdokumentation stimmt mit dem geplanten Einsatz überein
- UDI (Unique Device Identifier) auf Verpackung vorhanden und in EUDAMED abrufbar
- IT-Anforderungen (bei Software/digitalen Geräten) erfüllbar?
- Lagerungsanforderungen (Temperatur, Sterilität) mit Praxisbedingungen kompatibel
Haftungsrisiken bei nicht-konformen Produkten
Bei Patientenschäden durch nicht-konforme Produkte haften Sie als Betreiber ergänzend zum Hersteller, wenn Sie Ihre Sorgfaltspflicht (Prüfung von CE und Bestimmungsgemäßheit) verletzt haben. Verstöße gegen die Einweisungs- oder Dokumentationspflicht können als Organisationsverschulden gewertet werden, was im Prozess zur Beweislastumkehr zu Ihren Ungunsten führen kann.
Die Beweislastumkehr nach § 1 Abs. 4 ProdHaftG gilt für Hersteller — nicht für Betreiber. Im Arzthaftungsprozess trägt der Patient den Beweis, dass die fehlerhafte Produktanwendung den Schaden verursacht hat. Dennoch empfiehlt sich eine lückenlose Dokumentation der Beschaffungsentscheidung.
FAQ: Häufige Fragen zu unkritischen Medizinprodukten
Muss ich als Praxisinhaber die MDR-Konformität meiner Verbrauchsmaterialien aktiv prüfen?
Eine tiefgreifende Zertifizierungsprüfung müssen Sie nicht durchführen. Ihre Sorgfaltspflicht als Betreiber beschränkt sich auf die Sichtprüfung des CE-Kennzeichens, die Verfügbarkeit der Konformitätserklärung und die Überprüfung der Zweckbestimmung. Bei Produkten der Subklassen Is, Im und Ir müssen Sie zusätzlich prüfen, ob die vierstellige Kennnummer der Benannten Stelle hinter dem CE-Zeichen steht. Ab dem 28. Mai 2026 bietet die EUDAMED-Datenbank zudem eine verlässliche Quelle, um die Konformität neuer Produkte digital zu verifizieren und Fälschungen auszuschließen.
Was unterscheidet ein unkritisches Medizinprodukt von einem Alltagsgegenstand mit medizinischem Verwendungszweck?
Die Abgrenzung erfolgt über den vom Hersteller bestimmten Verwendungszweck (Artikel 2 Nr. 12 MDR). Ein Handschuh ohne medizinischen Verwendungszweck ist PSA (Persönliche Schutzausrüstung). Sobald der Hersteller das Produkt zur Anwendung am Patienten oder im medizinischen Kontext auslobt — z.B.„zur Untersuchung geeignet”— greift die MDR-Klassifizierungspflicht. Der Verwendungszweck ist in der Gebrauchsanweisung und der Konformitätserklärung dokumentiert.
Gilt die Betreiberpflicht nach MPDG auch für zugekaufte Klasse-I-Produkte ohne Messfunktion?
Ja, die Betreiberpflichten nach dem MPDG und der MPBetreibV gelten unabhängig von der Risikoklasse. Für einfacheKlasse-I-Produkte ohne Messfunktion entfällt in der Regel lediglich die Aufnahme in das Bestandsverzeichnis (§ 14 MPBetreibV).
Aber Vorsicht: Die allgemeine Einweisungspflicht wurde 2025 verschärft. Eine Einweisung des Personals ist nun bei jedem Produkt (auch Klasse I) verpflichtend, sofern eine Gebrauchsanweisung beiliegt. Die alte Ausnahme für „selbsterklärende“ Produkte ist damit weitestgehend hinfällig. Zudem bleiben die Pflicht zur bestimmungsgemäßen Verwendung und die Meldung von Vorkommnissen bestehen.