




Schneller Service
Kostenlose Rückmeldung innerhalb von 24 Stunden
Erfolg durch Erfahrung
Aus über 15.000 Projekten im Jahr wissen wir, worauf es ankommt
Der digitale Marktführer
Unsere Kunden sprechen für uns:
4,9 von 5 Sternen auf Google





Inhaltsverzeichnis
Was die MDR von Ärzten als Betreiber verlangt — und was nicht
Abgrenzung Hersteller- vs. Betreiberpflichten nach MDR Art. 2
Die MDR 2017/745 unterscheidet scharf zwischen Herstellern und Betreibern. Als Betreiber einer Arztpraxis sind Sie jede natürliche oder juristische Person, die ein Medizinprodukt in der Gesundheitsversorgung betreibt — also Ihre Praxis, Ihr MVZ oder Ihr ambulantes Operationszentrum. Herstellerpflichten wie die klinische Bewertung nach Anhang XIV oder die technische Dokumentation nach Anhang II berühren Sie nicht.
Was Sie betrifft, ist die sachgemäße Anwendung, Instandhaltung und Aufbereitung der eingesetzten Produkte. Diese Betreiberpflichten sind nicht in der MDR abschließend geregelt, sondern ergänzend in der Medizinprodukte-Betreiberverordnung (MPBetreibV). Beide Regelwerke greifen ineinander: Die MDR setzt den europäischen Rahmen, die MPBetreibV konkretisiert die nationalen Betreiberpflichten.
Haftungsrisiko: Betreiber, die Medizinprodukte ohne dokumentierte Risikobewertung aufbereiten, verstoßen gegen § 8 MPBetreibV. Im Schadensfall kann das als grob fahrlässige Pflichtverletzung gewertet werden.
Relevante Rechtsgrundlagen: MDR, MPBetreibV und RKI/KRINKO-Empfehlung im Zusammenspiel
Für die Risikobewertung von Medizinprodukten im Praxisalltag sind drei Rechtsquellen maßgeblich — keine davon darf isoliert betrachtet werden.
- Die MDR 2017/745 regelt in Anhang I die allgemeinen Sicherheits- und Leistungsanforderungen, denen aufbereitete Medizinprodukte genügen müssen.
- Die MPBetreibV verpflichtet Sie als Betreiber in § 8 zur Aufbereitung nach dem Stand der Technik — und benennt die KRINKO/BfArM-Empfehlung ausdrücklich als anerkannten Maßstab.
- Die RKI/KRINKO-Empfehlung (Bundesgesundheitsblatt 2012, 55:1244–1310) operationalisiert diesen Stand der Technik durch ein Drei-Stufen-Kategoriensystem.
Eine Aufbereitung gilt nach § 8 Abs. 2 MPBetreibV als ordnungsgemäß, wenn sie der KRINKO/BfArM-Empfehlung entspricht. Diese Konformitätsvermutung entlastet Sie im Zweifelsfall — setzt aber voraus, dass die Einstufung tatsächlich korrekt und schriftlich dokumentiert vorliegt (RKI/KRINKO 2012, Abschnitt 1.2.1).
MDR-Risikoklassen: Herstellerpflicht, Betreiberinformation
Die MDR 2017/745 verpflichtet Hersteller in Art. 51 i.V.m. Anhang VIII, jedes Medizinprodukt vor dem Inverkehrbringen einer Risikoklasse zuzuordnen. Es gibt vier Klassen: I, IIa, IIb und III, ergänzt durch die Sonderklassen Ir (wiederverwendbar) und Is (steril geliefert). Die Klassifizierung richtet sich nach Invasivität, Anwendungsdauer, Kontaktzone und spezifischen Produkteigenschaften (MDR Anhang VIII, Regeln 1–22). Je höher die Klasse, desto strenger das vorgeschriebene Konformitätsbewertungsverfahren — von der Selbstdeklaration des Herstellers bei Klasse I bis zur Beteiligung einer Benannten Stelle und klinischen Prüfung bei Klasse III (MDR Art. 52).
Als Betreiber müssen Sie die MDR-Klasse nicht selbst bestimmen. Sie lesen sie vom CE-Etikett oder der Konformitätserklärung des Herstellers ab. Was die Klasse für Sie auslöst, sind mittelbare Pflichten: Bei Klasse Ir muss der Hersteller eine Aufbereitungsanweisung nach DIN EN ISO 17664 bereitstellen — und Sie als Betreiber sind nach MPBetreibV § 8 verpflichtet, diese umzusetzen. Bei Klasse III-Implantaten greift die Rückverfolgbarkeitspflicht nach MDR Art. 27, und der Patient hat Anspruch auf einen Implantatausweis.
| MDR-Klasse | Risikostufe | Konformitätsbewertung | Typische Produkte | Betreiberrelevanz |
|---|---|---|---|---|
| I | Niedrig | Hersteller allein | Verbandmittel, Stethoskop, OP-Tisch | Aufbereitungsinfo nach DIN EN ISO 17664 prüfen |
| Ir | Niedrig, wiederverwendbar | Hersteller + Benannte Stelle (Aufbereitung) | Wiederverwendbare Chirurgieinstrumente, Küretten | Herstellerangaben zur Aufbereitung zwingend einzuhalten (MPBetreibV § 8) |
| Is | Niedrig, steril geliefert | Hersteller + Benannte Stelle (Sterilisation) | Steril verpackte Einwegprodukte | Verpackungsintegrität vor Anwendung prüfen |
| IIa | Mittel | Benannte Stelle erforderlich | Flexibles Endoskop, invasive Blutdruckmessung | Erhöhte Aufbereitungsanforderungen wahrscheinlich |
| IIb | Mittel-hoch | Benannte Stelle, strengere Prüfung | Beatmungsgerät, Röntgengerät, Langzeitimplantate | STK oder MTK nach MPBetreibV prüfen |
| III | Hoch | Benannte Stelle + ggf. klinische Prüfung | Koronarstents, Herzklappen, aktive Implantate | Rückverfolgbarkeit (MDR Art. 27); Implantatausweis für Patient |
Das Drei-Stufen-Modell: Einstufung nach RKI/KRINKO als Kernpflicht
Unkritisch, semikritisch, kritisch: Kriterien und Abgrenzung
Die Risikoeinstufung nach RKI/KRINKO orientiert sich ausschließlich an der Art der Patientenexposition — nicht am Materialwert, der Komplexität oder dem Anschaffungspreis des Instruments. Entscheidend ist, mit welchen Körperbereichen das Produkt bestimmungsgemäß in Kontakt kommt.
Unkritisch sind Medizinprodukte, die lediglich mit intakter Haut in Berührung kommen — etwa EKG-Elektroden oder Blutdruckmanschetten. Eine Desinfektion der äußeren Oberfläche ist ausreichend, Sterilisation ist nicht erforderlich.
Semikritisch sind Medizinprodukte mit Kontakt zu Schleimhaut oder krankhaft veränderter Haut — zum Beispiel Specula oder flexible Endoskope. Hier ist mindestens eine Desinfektion mit einem Wirkungsspektrum erforderlich, das Bakterien einschließlich Mykobakterien, Pilze und Viren erfasst (RKI/KRINKO 2012, Tab. 1). Semikritische Produkte werden in A (ohne erhöhte Aufbereitungsanforderungen) und B (mit erhöhten Anforderungen, z. B. flexible Endoskope) unterteilt.
Kritisch sind Medizinprodukte, die Haut oder Schleimhaut durchdringen und dabei mit Blut, inneren Geweben oder Organen in Kontakt kommen — chirurgische Instrumente, Trokar-Systeme. Sie müssen sterilisiert werden. Kritische Produkte werden weiter in die Untergruppen A, B und C differenziert, je nach Aufbereitungsaufwand und thermischer Stabilität.
Risikobewertung Medizinprodukte — Tabelle: RKI/KRINKO-Kategorien mit Aufbereitungsanforderungen
| Kategorie | Kontaktbereich | Beispiele | Desinfektion | Sterilisation | Besonderheiten |
|---|---|---|---|---|---|
| Unkritisch | Intakte Haut | EKG-Elektrode, Blutdruckmanschette | Flächendesinfektion | Nein | — |
| Semikritisch A | Schleimhaut / veränd. Haut | Spekulum, Laryngoskopspatel | Ja (bakterizid, fungizid, viruzid) | Nein (fakultativ) | — |
| Semikritisch B | Schleimhaut, komplexe Geometrie | Flexibles Gastroskop, Bronchoskop | Ja, bevorzugt maschinell | Nein (fakultativ) | Validierungspflicht, KRINKO Anlage 8 (2024) |
| Kritisch A | Penetration, unkompliziert | Wundhaken, einfache Chirurgieinstrumente | Ja, bevorzugt maschinell | Ja (feuchte Hitze) | — |
| Kritisch B | Penetration, erhöhte Anforderungen | MIC-Trokare, Laparoskopie-Instrumente | Ja, maschinell (RDG) | Ja (feuchte Hitze) | Sachkundenachweis erforderlich |
| Kritisch C | Penetration, thermolabil | Thermolabile Laparoskope, bestimmte Neurochirurgie-Instrumente | Ja, maschinell | Ja (Niedertemperaturverfahren) | Externe QM-Zertifizierungspflicht |
Zweifelsfälle: Das Gebot der höheren Risikostufe
Bei Zweifeln an der Einstufung ist das Medizinprodukt der höheren, also kritischeren Risikostufe zuzuordnen (RKI/KRINKO 2012, Abschnitt 1.2.1). Dieses Gebot der Vorsicht gilt bei Instrumenten, die konstruktionsbedingt schwer zu reinigen sind — etwa wegen langer, enger Lumina, Hohlräumen mit nur einer Öffnung oder schlecht zugänglicher Oberflächen. Auch Instrumente mit elektronischen Anteilen oder knicksensiblen Oberflächen erfordern eine differenzierte Einzelbetrachtung.
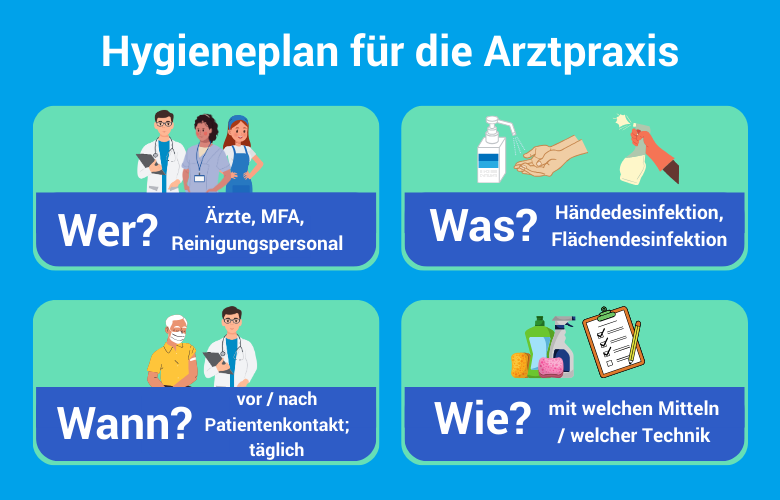
Tipp: Tipp: Führen Sie bei unklarer Einstufung eine kurze schriftliche Begründung im Produktdossier. Das belegt im Prüfungsfall, dass Sie sich aktiv mit der Risikobewertung auseinandergesetzt haben. Diese Dokumentation sollte nahtlos in Ihr einrichtungsinternes Hygienemanagement zur Infektionsprävention einfließen, um bei Begehungen durch das Gesundheitsamt eine lückenlose Argumentationskette vorweisen zu können.
Risikobewertung Medizinprodukte in der Arztpraxis: Schritt für Schritt
Produkterfassung und Dokumentationspflicht nach MPBetreibV
Der erste operative Schritt ist die vollständige Bestandsaufnahme aller aufbereitungsrelevanten Medizinprodukte in Ihrer Praxis. Für jedes Produkt — oder jede Produktfamilie mit identischen Aufbereitungsanforderungen — ist schriftlich festzulegen, ob und mit welchen Verfahren es aufbereitet wird (RKI/KRINKO 2012, Abschnitt 1.2.1). Diese Risikobewertung bildet die Grundlage für die weiteren konkreten Anforderungen an die Instrumentenaufbereitung, insbesondere im Hinblick auf die Validierung der Reinigungs- und Desinfektionsprozesse.
Die Dokumentation umfasst nach MPBetreibV mindestens folgende Elemente:
- Produktbezeichnung, Artikelnummer, Hersteller
- Risikoklassifikation nach RKI/KRINKO (mit Begründung bei Zweifelsfällen)
- Festgelegtes Aufbereitungsverfahren (Reinigungs-, Desinfektions-, ggf. Sterilisationsverfahren)
- Verweis auf Herstellerangaben zur Aufbereitung (DIN EN ISO 17664)
- Requalifizierungserfordernis bei Produktwechsel
Die Aufzeichnungen müssen während der Aufbewahrungsfrist jederzeit verfügbar und lesbar sein — und auf Verlangen den zuständigen Behörden vorgelegt werden (MPBetreibV § 9).
Herstellerangaben einbeziehen: DIN EN ISO 17664 als Ausgangspunkt
Bevor Sie eigenständig eine Einstufung vornehmen, müssen Sie die Angaben des Herstellers berücksichtigen — die MPBetreibV schreibt das ausdrücklich vor. Die DIN EN ISO 17664 verpflichtet Hersteller, für jedes aufbereitbare Medizinprodukt detaillierte Informationen zu Reinigung, Desinfektion und Sterilisation bereitzustellen. Diese Informationen sind Ausgangspunkt Ihrer eigenen Risikobewertung, nicht deren Ersatz.
Prüfen Sie insbesondere: Macht der Hersteller Angaben zur maximalen Anzahl von Aufbereitungszyklen? Bestehen Einschränkungen hinsichtlich bestimmter Sterilisationsverfahren? Liegen solche Angaben nicht vor oder sind unvollständig, ist das ein relevantes Risikosignal — und erhöht Ihren eigenen Dokumentationsaufwand (RKI/KRINKO 2012, Abschnitt 1.2).
Risikobewertung Medizinprodukte — Tabelle: Muster-Bewertungsliste für Arztpraxis und Zahnarztpraxis
| Produkt | Art.-Nr. / Hersteller | Normative Anforderung | KRINKO-Einstufung | Aufbereitungsverfahren | Requalifizierung bei Wechsel? |
|---|---|---|---|---|---|
| Wundhaken, gerade | [individuell] | MDR Anhang I; DIN EN ISO 17664 | Kritisch A | Maschinell RDG + Dampfsterilisation 134 °C | RDG-Prozess, Sterilisationsprozess |
| Spezialkürette (Büro-Eingriff) | [individuell] | MDR Anhang I; DIN EN ISO 17664 | Kritisch A | Maschinell RDG + Dampfsterilisation | Sterilisationsprozess |
| Laryngoskopspatel (Metall) | [individuell] | MDR Anhang I | Semikritisch A | Thermische Desinfektion RDG | RDG-Prozess |
| Reinigungsbürste für Lumina | In der Regel kein MP | Keine spez. Vorgabe | — (Hilfsmittel) | Prozesseignung nachweisen | Reinigungsleistung prüfen |
| Prozesschemikalie RDG | MDR Anhang I | KRINKO/BfArM-konform; Materialverträglichkeit | — (Hilfsstoff) | Herstellerfreigabe prüfen | Reinigungs-/Desinfektionsprozess |
Risikobewertung Medizinprodukte in der Zahnarztpraxis: Besonderheiten
Semikritisch B: Zahnärztliches Instrumentarium mit erhöhten Anforderungen
Die Zahnarztpraxis unterscheidet sich von einer allgemeinmedizinischen Praxis durch das systematische Vorkommen von Instrumenten der Kategorie semikritisch B und kritisch B. Zahnärztliche Hand- und Winkelstücke, Ultraschallansätze mit Innenkanälen oder Spüllösungskanäle stellen wegen ihrer komplexen Geometrie besondere Anforderungen an die Aufbereitung.
Für semikritisch B klassifizierte Instrumente gilt die bevorzugte maschinelle Reinigung und Desinfektion im validierten Reinigungs- und Desinfektionsgerät (RDG) als verbindlicher Stand der Technik. Die KRINKO hat in der Neufassung der Anlage 8 (Bundesgesundheitsblatt 2024, S. 1410–1468) klargestellt, dass die Aufbereitung dieser Produkte nur in speziell ausgestatteten Einheiten erfolgen darf, die über die notwendige technische Infrastruktur zur Validierung der Kanalreinigung verfügen.
Kritisch C: Thermolabile Instrumente — externe Zertifizierungspflicht
Thermolabile Instrumente, die bestimmungsgemäß Gewebe penetrieren und nicht bei 134 °C dampfsterilisierbar sind, sind der Kategorie kritisch C zuzuordnen (RKI/KRINKO 2012, Abschnitt 1.2.1). Die Aufbereitung durch Niedertemperatursterilisationsverfahren — z. B. VH₂O₂-Plasma — erfordert die Zertifizierung des Qualitätsmanagementsystems (QM) in der Arztpraxis bzw. QM Zahnarztpraxis durch eine externe Stelle, in der Regel nach DIN EN ISO 13485. Diese Pflicht entfällt nur, wenn der Hersteller ein bestimmtes alternatives Verfahren benennt und dieses vor Ort validiert wurde.
Haftungsrisiko: Kritisch C-Produkte ohne validierten Prozess und externe QM-Zertifizierung dürfen nicht aufbereitet und wiederverwendet werden. Eine Eigendeklaration reicht nicht aus.
Einwegprodukte vs. Mehrweginstrumente: Wann ist Wiederaufbereitung zulässig?
Ist ein Medizinprodukt vom Hersteller als Einwegprodukt gekennzeichnet, ist die Wiederaufbereitung nach MDR Art. 17 nur unter engen Bedingungen zulässig. Wer ein Einwegprodukt aufbereitet, um es erneut einzusetzen, gilt nach MDR Art. 17 Abs. 2 als Hersteller des aufbereiteten Produkts — mit allen daraus resultierenden Herstellerpflichten. Für niedergelassene Praxen ist dieser Weg praktisch nicht gangbar, da Konformitätsbewertung und Haftungsübernahme die Kapazitäten einer Einzelpraxis regelmäßig übersteigen.
Die Risikobewertung von Medizinprodukten muss daher bereits bei der Beschaffungsentscheidung beginnen: Ist ein Instrument als Einwegprodukt konzipiert, ist der Aufbereitungsweg von vornherein verschlossen — unabhängig von Preis oder scheinbarer Robustheit.
Nicht-Medizinprodukte im Praxisalltag: Was gilt für Instrumentarium ohne CE-Kennzeichnung?
VAH/KPH-Empfehlung: Risikoeinstufung analog zu Medizinprodukten
Im Praxisalltag begegnen Ihnen regelmäßig Instrumente und Arbeitsgeräte, die rechtlich nicht als Medizinprodukt eingestuft sind — etwa bestimmte Reinigungsbürsten, Lagerungshilfsmittel oder Prozesschemikalien. Für solche Produkte gibt es keinen expliziten gesetzlichen Einstufungsrahmen, der mit der KRINKO-Empfehlung vergleichbar wäre.
Die Kommission für Praktische Hygiene im VAH empfiehlt in ihrer Publikation vom Dezember 2025, das Risikobewertungsschema der KRINKO/BfArM-Empfehlung analog anzuwenden — also eine Einstufung nach dem potenziellen Kontakt mit Patienten oder übertragungsrelevanten Körperflüssigkeiten vorzunehmen. Für aufbereitungsrelevante Hilfsstoffe wie Prozesschemikalien verlangt die DGSV-Empfehlung Nr. 127 zudem einen Nachweis der Prozesseignung und — soweit rückstandsrelevant — der Biokompatibilität (FA Qualität Empfehlung Nr. 127, Zentralsterilization 1/2024).
Vergleich: Medizinprodukt vs. nicht-medizinisches Instrumentarium
| Kriterium | Medizinprodukt (CE-gekennzeichnet) | Nicht-medizinisches Instrumentarium |
|---|---|---|
| Rechtsgrundlage | MDR 2017/745, MPBetreibV | Keine spez. Norm; Analogieanwendung empfohlen |
| Risikoklassifikation | Pflicht nach RKI/KRINKO (schriftlich) | Empfohlen nach VAH/KPH-Schema |
| Herstellerangaben | Verpflichtend (DIN EN ISO 17664) | Produktdatenblatt und Eignungsnachweis vorhalten |
| Aufbereitungsdokumentation | Pflicht (MPBetreibV § 9) | Best practice; erwartet bei Behördenprüfung |
| Biokompatibilitätsnachweis | Durch Hersteller (MDR Anhang I) | Herstellernachweis bei Rückstandsrisiko |
| Konsequenz bei Verstoß | Haftung nach MPBetreibV, ggf. Strafrecht | Haftung nach allg. Deliktsrecht (§ 823 BGB) |
Dokumentation und Qualitätssicherung: Was muss schriftlich vorliegen?
Risikoanalyse, Validierungsberichte und Schulungsnachweise
Die Risikobewertung von Medizinprodukten ist kein einmaliger Akt — sie ist ein fortlaufender Prozess. MPBetreibV und KRINKO-Empfehlung benennen konkrete Dokumentationselemente, die bei einer Behördenprüfung oder im Schadensfall vorzulegen sind.
Das Betreiberdokument zur Risikobewertung enthält die Einstufung jedes Produkts nach KRINKO-Kategorien, die Begründung bei Zweifelsfällen sowie die Festlegung der Aufbereitungsverfahren. Es bildet die Grundlage für alle weiteren Qualifizierungsdokumente.
Der Validierungsbericht des Reinigungs- und Desinfektionsgeräts sowie des Sterilisators belegt die Eignung der eingesetzten Verfahren. Er umfasst Installationsqualifikation (IQ), Betriebsqualifikation (OQ) und Leistungsqualifikation (PQ). Chargenprotokolle dokumentieren jeden Aufbereitungszyklus und ermöglichen die Rückverfolgbarkeit.
Schulungsnachweise für das mit der Medizinprodukte-Aufbereitung betraute Personal sind nach RKI/KRINKO für kritisch B-Produkte explizit vorgeschrieben. Zu dokumentieren sind Schulungsinhalt, Teilnehmer, Unterrichtender und Datum (RKI/KRINKO 2012, Anlage 4).
Aufbewahrungsfristen und Behördenzugang nach MPBetreibV
Die MPBetreibV schreibt vor, dass alle Aufzeichnungen jederzeit verfügbar und lesbar sein müssen — auf Papier oder elektronischen Datenträgern. Änderungen dürfen den ursprünglichen Inhalt nicht unkenntlich machen; Korrekturen müssen als solche erkennbar sein. Nach Behördenangaben wird für Chargendokumentation eine Aufbewahrung von mindestens 5 Jahren, aus Haftungsgründen von bis zu 20 Jahren empfohlen. Auf Verlangen der zuständigen Landesbehörde sind alle Unterlagen vorzulegen — Validierungsberichte, Chargenprotokolle und Schulungsdokumentation eingeschlossen (MPBetreibV § 9 Abs. 2).
Tipp: Führen Sie Chargenprotokolle digital und systemgestützt, sofern Ihr Sterilisator das erlaubt. Das reduziert den manuellen Aufwand und gewährleistet lückenlose Rückverfolgbarkeit ohne Zusatzaufwand.
FAQ: Häufige Fragen zur Risikobewertung von Medizinprodukten
Muss ich für jedes Medizinprodukt eine eigene Risikobewertung erstellen?
Nicht zwingend für jedes einzelne Instrument. Die KRINKO-Empfehlung lässt die Zusammenfassung zu Produktfamilien zu, sofern die Produkte identische aufbereitungsrelevante Eigenschaften aufweisen (RKI/KRINKO 2012, Abschnitt 1.2.1). Bei abweichenden Geometrien, Materialien oder Herstellervorgaben ist eine Einzelbewertung erforderlich. Entscheidend ist stets, dass die Einstufung und das Aufbereitungsverfahren für jede Gruppe vollständig dokumentiert sind.
Was passiert, wenn mein Produkt nicht eindeutig einer KRINKO-Kategorie zuzuordnen ist?
Die Vorgabe ist eindeutig: Im Zweifelsfall ordnen Sie das Produkt der höheren Risikostufe zu (RKI/KRINKO 2012, Abschnitt 1.2.1). Diese Entscheidung ist schriftlich zu begründen und im Betreiberdokument festzuhalten. Bei komplexen Fällen ziehen Sie frühzeitig den zuständigen Hygieniker oder einen Aufbereitungsexperten hinzu — das ist fachlich geboten und haftungsrechtlich klug.
Gilt die Risikobewertungspflicht auch für zugekaufte Einwegprodukte?
Für Einwegprodukte, die nach einmaliger Anwendung entsorgt werden, entfällt die Aufbereitungsdokumentation. Gleichwohl sollten Sie schriftlich festhalten, dass das Produkt als Einwegprodukt eingestuft wurde und welche Entsorgungsroute gewählt wird. Für Einwegprodukte, die als Hilfsmittel im Aufbereitungsprozess eingesetzt werden — z. B. Einwegverpackungen für steriles Sterilgut —, gelten die Anforderungen der DIN EN ISO 11607 (FA Qualität Empfehlung Nr. 127, Zentralsterilization 1/2024).