



Schneller Service
Kostenlose Rückmeldung innerhalb von 24 Stunden
Erfolg durch Erfahrung
Aus über 15.000 Projekten im Jahr wissen wir, worauf es ankommt
Der digitale Marktführer
Unsere Kunden sprechen für uns:
4,9 von 5 Sternen auf Google





Abstract – Reinigung, Desinfektion und Sterilisation in der Arztpraxis
- Die drei Aufbereitungsverfahren verfolgen unterschiedliche Ziele: Reinigung entfernt organische Verunreinigungen (ohne Keimwirkung), Desinfektion reduziert vegetative Mikroorganismen auf ein definiertes Maß, Sterilisation inaktiviert alle vermehrungsfähigen Keime inklusive Sporen bis zu einem SAL ≤ 10⁻⁶.
- Die KRINKO/BfArM-Empfehlung stuft Medizinprodukte in fünf Risikoklassen ein (unkritisch, semikritisch A/B, kritisch A/B/C); die Klassifikation bestimmt das vorgeschriebene Aufbereitungsverfahren – eine Fehlzuordnung begründet im Schadensfall eine Haftung nach MPDG und BGB mit Beweislastumkehr gemäß § 630h Abs. 4 BGB.
- Für kritische Medizinprodukte mit Hohlräumen (Klasse B) ist ausschließlich ein Klasse-B-Autoklav mit fraktioniertem Vorvakuum zulässig; Klasse-N-Geräte sind für verpackte oder hohle Instrumente ungeeignet und dürfen für diese nicht eingesetzt werden.
- Lückenlose Chargendokumentation (empfohlene Aufbewahrungsdauer: 30 Jahre) und aktuelle Validierungsberichte (IQ, OQ, jährliche PQ nach DIN EN ISO 17665) sind keine optionalen Ergänzungen, sondern die einzige rechtssichere Entlastung im Haftungsfall.
Inhaltsverzeichnis
Was bedeuten Reinigung, Desinfektion und Sterilisation — und wie grenzen sie sich ab?
Die Aufbereitung von Medizinprodukten erfolgt in konsekutiven Prozessschritten mit spezifischem Zielspektrum. Maßgeblich ist die gemeinsame Empfehlung der KRINKO und des BfArM, welche die normative Basis bilden.
- Reinigung bezeichnet die Entfernung von sichtbaren Verunreinigungen, Blut, Proteinen und organischem Material von einer Oberfläche. Sie tötet keine Mikroorganismen ab, schafft aber die Voraussetzung dafür, dass nachfolgende Desinfektions- oder Sterilisationsverfahren überhaupt wirken können. Organische Rückstände auf einem Instrument blockieren chemische Wirkstoffe und schützen Erreger vor thermischer Inaktivierung.
- Desinfektion zielt auf die Reduktion oder Abtötung krankheitserzeugender Mikroorganismen. Das Ergebnis ist keine Keimfreiheit, sondern eine definierte Keimreduktion — abhängig vom eingesetzten Verfahren und dem Wirkspektrum des Desinfektionsmittels. Bakteriensporen bleiben hierbei meist unbeeinträchtigt.
- Sterilisation finalisiert die Aufbereitung. Ziel ist die Inaktivierung aller vermehrungsfähigen Mikroorganismen inklusive hochresistenter Sporen. Der normative Zielwert ist ein Sterility Assurance Level (SAL) von ≤ 10⁻⁶. Die Validierung erfolgt primär auf Basis der DIN EN ISO 17665 (Dampfsterilisation) bzw. für Kleinsterilisatoren nach DIN EN 13060.
| Verfahren | Ziel | Wirkspektrum | Verbleibende Keime |
| Reinigung | Entfernung organischer Verunreinigungen | Keines (mechanisch) | Alle Mikroorganismen möglich |
| Desinfektion | Keimreduktion | Vegetative Bakterien, Viren | Sporen häufig resistent |
| Sterilisation | Vollständige Keimfreiheit | Alle Mikroorganismen inkl. Sporen | Keine (SAL ≤ 10⁻⁶) |
Wie klassifiziert das RKI Medizinprodukte — und welches Verfahren folgt daraus?
Die KRINKO-Empfehlung unterscheidet drei Hauptkategorien, die sich aus dem Kontaktrisiko des Medizinprodukts mit dem Patienten ableiten. Dabei ist die Risikobewertung kein administrativer Akt, sondern ein strategisches Risikomanagement im Rahmen der gesamten Instrumentenaufbereitung gemäß RKI.
Unkritisch, semikritisch, kritisch: die drei Risikoklassen
- Unkritische Medizinprodukte: Kontakt nur mit intakter Haut (z. B. Blutdruckmanschetten). Reinigung und ggf. Wischdesinfektion genügen.
- Semikritische Medizinprodukte (A/B): Kontakt mit Schleimhaut oder nicht-intakter Haut. Semikritisch A (ohne besondere Anforderungen an die Aufbereitung z. B. Spatel), in der Regel genügt Desinfektion. Semikritisch B (z. B. flexible Endoskope) erfordert aufgrund der Komplexität immer eine maschinelle, validierte Aufbereitung.
- Kritische Medizinprodukte (A/B/C): Diese Produkte penetrieren Haut oder Schleimhaut. Hier ist die Sterilisation zwingend. Kritisch B (Hohlkörper wie Spülkanülen) verlangt zwingend einen Klasse-B-Autoklaven mit fraktioniertem Vorvakuum. Kritisch C umfasst thermolabile Produkte, die oft Niedertemperaturverfahren (Plasma) benötigen.
In der Praxis des Jahres 2026 hat sich die Differenzierung innerhalb der Kategorien weiter verfeinert. Die Unterscheidung zwischen semikritisch A und B sowie kritisch A, B und C orientiert sich stark an der mechanischen Komplexität und der Thermostabilität der Instrumente.
| Risikogruppe | Definition und Kontakt | Aufbereitungsziel | Beispielhaftes Instrumentarium |
| Unkritisch | Kontakt nur mit intakter Haut | Reinigung, ggf. Wischdesinfektion | Blutdruckmanschetten, EKG-Elektroden, Stethoskope |
| Semikritisch A | Kontakt mit Schleimhaut oder nicht intakter Haut; keine erhöhten Anforderungen an die Reinigung | Reinigung und Desinfektion (thermisch bevorzugt) | Spekula, Zungenspatel, Mundspiegel |
| Semikritisch B | Kontakt mit Schleimhaut oder nicht intakter Haut, aber mit erhöhten Anforderungen (Hohlkörper, schwer zugängliche Stellen) | Reinigung und validierte Desinfektion (maschinell obligat) | Flexible Endoskope ohne Biopsiekanal, Ultraschallsonden mit Schleimhautkontakt |
| Kritisch A | Anwendung von Blut, Gewebe oder sterilen Arzneimitteln; keine erhöhten Anforderungen | Reinigung, Desinfektion, Sterilisation | Skalpelle, Wundhaken, Pinzetten |
| Kritisch B | Anwendung von Blut, Gewebe oder sterilen Arzneimitteln, aber mit erhöhten Anforderungen (Hohlkörper, Lumen) | Validierte Reinigung/Desinfektion, Sterilisation (Klasse B) | Chirurgische Handstücke, Spülkanülen. Biopsiezangen |
| Kritisch C | Anwendung von Blut, Gewebe oder sterilen Arzneimitteln, aber thermolabil (nicht 134°C fest) | Reinigung/Desinfektion, Niedertemperatur-Sterilisation | Thermolabile Endoskope für sterile Eingriffe, spezielle Optiken |
Haftungsrisiko: Eine Fehlklassifikation indiziert bei Infektionen eine Haftung nach dem Medizinprodukterecht-Durchführungsgesetz (MPDG) sowie dem BGB. Besonders kritisch: Bei Verstößen gegen RKI-Richtlinien (anerkannte Regeln der Technik) greift die Beweislastumkehr gemäß § 630h Abs. 4 BGB. In diesem Fall müssen Sie beweisen, dass die Infektion nicht auf Ihrer Aufbereitung beruht.
Reinigung: Voraussetzung jeder weiteren Aufbereitung
Manuelle vs. maschinelle Reinigung
Reinigung ist der limitierende Schritt der gesamten Aufbereitungskette: Kein Desinfektions- und kein Sterilisationsverfahren kompensiert eine unzureichende Vorreinigung. Protein- und Blutrückstände auf Instrumenten reduzieren die Wirksamkeit thermischer Verfahren messbar — und können bei chemischer Desinfektion die Reaktivität des Wirkstoffs vollständig neutralisieren.
Die manuelle Reinigung unter fließendem Wasser mit Reinigungsbürste und enzymhaltigen Reiniger ist für einfache, unstrukturierte Instrumente akzeptabel. Für Hohlkörper, Gelenkinstrumente und komplexe Instrumente schreibt die KRINKO jedoch die maschinelle Reinigung im Reinigungs- und Desinfektionsgerät (RDG) vor, da eine reproduzierbare manuelle Reinigung dieser Strukturen nicht validierbar ist.
Das RDG gewährleistet einen standardisierten, maschinellen Prozess gemäß DIN EN ISO 15883. Der A₀-Wert (Zeit-Temperatur-Relation) muss für Instrumente mit viruzider Anforderung i. d. R. ≥ 3000 betragen (z. B. 90 °C für 5 Min.), während für vegetative Keime ≥ 600 ausreichen. Die maschinelle Aufbereitung ist aufgrund der Validierbarkeit der manuellen Aufbereitung gemäß der KRINKO-Empfehlung vorzuziehen.
Desinfektion: Verfahren, Wirkspektren und Grenzen
Thermische vs. chemische Desinfektion
Die thermische Desinfektion — im RDG bei ≥ 90 °C für definierte Zeiten — ist der chemischen Desinfektion grundsätzlich vorzuziehen: Sie ist reproduzierbar, validierbar und hinterlässt keine toxischen Rückstände auf Instrumenten. Ihr Einsatz setzt jedoch thermostabile Instrumente voraus.
Die chemische Desinfektion ist für thermolabile Produkte oder Flächen indiziert. Entscheidend ist die Auswahl nach gelistetem Wirkspektrum: Die VAH-Liste (Verbund für angewandte Hygiene) und die DVG-Liste (Deutsche Veterinärmedizinische Gesellschaft) führen Präparate nach nachgewiesenem Wirkspektrum.
Für die Routineaufbereitung in der Arztpraxis sind folgende Parameter von Bedeutung: Der A₀-Wert von 600 gilt als Standard für die Inaktivierung vegetativer Bakterien und behüllter Viren (HBV, HIV, HCV) bei einer Temperatur von 80°C über 600 Sekunden oder 90°C über 60 Sekunden.
Die maschinelle Aufbereitung im RDG nach DIN EN ISO 15883 stellt sicher, dass diese Parameter in jedem Zyklus reproduzierbar eingehalten werden, was bei manuellen Verfahren faktisch nicht nachweisbar ist.
Grenzen der Desinfektion: Desinfektion erreicht keine Sterilität. Für alle kritischen Medizinprodukte ist sie daher nicht als abschließendes Verfahren zugelassen.
Besonders relevant: Prionen-Schutz – Standarddesinfektion wirkt nicht gegen Prionen (CJK-Übertragung). Hier sind spezifische Sterilisationsparameter (134°C, 18 Min.) nötig.
Sterilisation: Verfahren und Anforderungen für die Arztpraxis
Dampfsterilisation, Heißluft, Plasma
Die Dampfsterilisation im Autoklav ist das Standardverfahren für thermostabile Medizinprodukte in der Arztpraxis. Die DIN EN 13060 klassifiziert Kleinsterilisatoren in drei Klassen:
- Klasse B: Universell einsetzbar für verpackte Produkte, Hohlkörper und poröse Güter. Pflicht für alle kritischen B- und C-Instrumente.
- Klasse S: Für spezifizierte Produktgruppen — der Hersteller des Sterilisators muss exakt definieren, für welche Produkte das Gerät zugelassen ist.
- Klasse N: Nur für unverpackte, massiv-solide Instrumente ohne Hohlräume oder poröse Güter. Für kritische Medizinprodukte absolut unzulässig.
Die Heißluftsterilisation (180 °C, 30 min) ist für Instrumente mit Verpackung kaum noch relevant, da sie Verpackungsmaterialien beschädigt und der Dampfsterilisation in Effizienz und Sicherheit unterlegen ist.
Die Plasmasterilisation (Niedertemperatur-H₂O₂-Gas-Plasma) ist für thermolabile Optiken und Elektronik geeignet. Ermöglicht eine Sterilisation zwischen 45°C und 55°C.
Hinweis: Sterilisatoren der Klasse N sind ausschließlich für unverpackte, massive Instrumente (nicht-kritisch) konzipiert. Da sie über kein fraktioniertes Vorvakuum verfügen, ist eine Entlüftung von Hohlkörpern oder die Durchdringung von Sterilbarrieresystemen (Verpackungen) physikalisch nicht gewährleistet. Der Einsatz für kritische Medizinprodukte stellt einen schwerwiegenden Hygienemangel dar.
Erfahren Sie hier im Detail, wie das Funktionsprinzip der Sterilisation im Autoklaven funktioniert.
Validierungspflicht
Jeder Sterilisator benötigt eine Installations- (IQ), Betriebs- (OQ) und eine jährliche Leistungsqualifikation (PQ) nach DIN EN ISO 17665:2024. Ohne aktuelle PQ gilt der Prozess rechtlich als nicht validiert.
Die Routineprüfung umfasst:
- Bowie-Dick-Test: (täglich vor dem ersten Sterilisationszyklus bei Klasse-B-Geräten): Prüft die Dampfdurchdringung poröser Güter.
- Helix-Test: (alternativ zum Bowie-Dick-Test): Prüft Dampfdurchdringung von Hohlkörpern.
- Sporentest: (biologische Prüfung nach Risikoprofil): Weist die tatsächliche Abtötung hochresistenter Bakteriensporen als direkten Wirksamkeitsnachweis nach.
- Vakuumtest: (täglich vor dem ersten Zyklus bei Klasse-B-Geräten): Prüft die mechanische Dichtigkeit der Druckkammer durch Messung der Leckrate.
- Chargendokumentation: Jede Charge mit Datum, Charge-Nr., Programm, Ergebnis, Freizeichnung
Dokumentations- und Hygienepflichten in der Arztpraxis
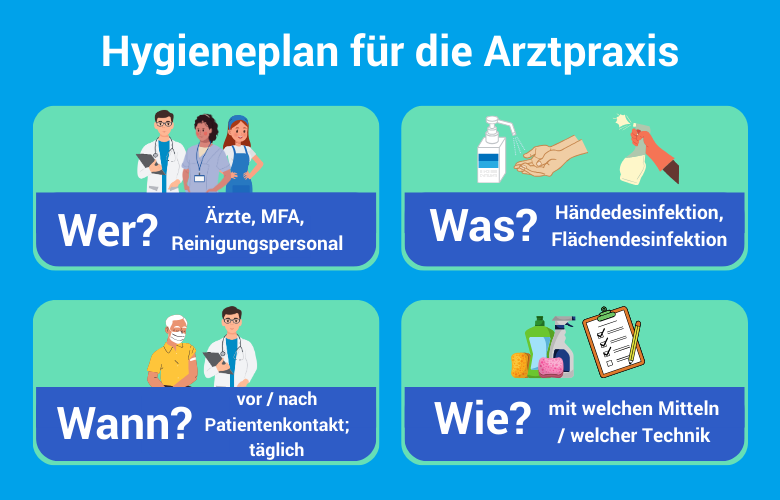
Hygieneplan und Rückverfolgbarkeit
§ 23 Abs. 3 IfSG verpflichtet Arztpraxen zur Erstellung und Einhaltung eines Hygieneplans. Dieser muss die eingesetzten Aufbereitungsverfahren, Verantwortlichkeiten und Prüfintervalle verbindlich festlegen. Die Prüfpflicht durch das Gesundheitsamt ist in den meisten Bundesländern konkretisiert durch Hygieneverordnungen auf Landesebene.
Die Chargendokumentation ist keine optionale Ergänzung, sondern ein Pflichtbestandteil der validierten Aufbereitung. Im Schadensfall müssen Sie nachweisen können, mit welchem validierten Verfahren ein Instrument aufbereitet wurde, das bei einem bestimmten Patienten eingesetzt wurde. Ohne lückenlose Dokumentation ist dieser Nachweis nicht möglich.
- Hygieneplan schriftlich dokumentiert und jährlich aktualisiert
- Aufbereitungsverantwortlicher benannt
- Produktklassifikation für alle eingesetzten Medizinprodukte dokumentiert
- Validierungsnachweise für RDG und Autoklav vorhanden
- Chargendokumentation für jeden Sterilisationszyklus
- Bowie-Dick-/Helix-Testergebnisse archiviert (Aufbewahrung mind. 5 Jahre, Empfohlen 30 Jahre § 199 BGB)
- VAH/DVG-gelistete Desinfektionsmittel mit Einwirkzeiten im Hygieneplan hinterlegt
Rechtssicherheit durch Dokumentation: Die lückenlose digitale oder analoge Chargendokumentation ist die einzige Entlastung im Haftungsprozess. Gemäß § 630h BGB führt das Fehlen dokumentierter Prozessparameter zur Vermutung der Fehlerhaftigkeit. Eine rechtssichere Dokumentation muss die Freigabeentscheidung durch eine sachkundige Person sowie die physischen Prozessparameter (Druck, Temperatur, Zeit) beinhalten.
Dokumentations-Checkliste
| Dokumentengruppe | Erforderlicher Inhalt / Nachweis | Archivierungsfrist (Empfehlung) |
| Hygieneplan | Aktuelle Version (Stand 2025/2026), schriftliche Benennung von Verantwortlichkeiten, Reinigungs- und Desinfektionspläne. | Fortlaufend, mind. 10 J. |
| Produktklassifizierung | Liste aller Instrumente mit Zuordnung zu RKI-Klassen und Aufbereitungsverfahren. | Bis Außerbetriebnahme + 10 J. |
| Validierungsberichte | IQ, OQ und aktuelle PQ (nicht älter als 12 Monate) für RDG und Autoklav. | Gesamte Lebenszeit des Geräts. |
| Chargendokumentation | Prozessprotokolle jedes Zyklus inkl. Freigabe durch Name/Signatur, Datum, Chargennummer. | 30 Jahre (Haftungsschutz). |
| Routineprüfungen | Tägliche Bowie-Dick- oder Helix-Testergebnisse, Vakuumtestprotokolle. | 30 Jahre. |
| Personalqualifikation | Sachkundenachweise (BÄK/DGSV), Einweisungsbelege nach MPBetreibV, Fortbildungszertifikate. | Dauer der Beschäftigung + 10 J. |
| Medizinproduktebuch | Bestandsverzeichnis, Wartungshistorie, sicherheitstechnische Kontrollen (STK). | 5 J. nach Außerbetriebnahme. |
FAQ: Häufige Fragen zu Reinigung, Desinfektion und Sterilisation
Müssen unkritische Medizinprodukte zwingend desinfiziert werden?
Ja, bei direktem Patientenkontakt (z. B. EKG-Elektroden) ist eine desinfizierende Aufbereitung nach KRINKO-Standard vorgeschrieben.
Was unterscheidet Klasse-B- von Klasse-S-Autoklaven in der Praxis?
Klasse-B-Geräte sind universell einsetzbar für verpackte und unverpackte, solide und hohle, poröse und nicht-poröse Güter. Klasse-S-Geräte sind auf ein vom Hersteller definiertes Produktspektrum beschränkt — Hohlkörper oder poröse Güter können außerhalb dieser Spezifikation nicht steril aufbereitet werden. Wer ein Klasse-S-Gerät für Instrumente einsetzt, die nicht im Validierungsumfang enthalten sind, verlässt den validierten Prozess und haftet bei Schadenseintritt vollumfänglich.
Welche Konsequenzen drohen bei fehlender Validierungsdokumentation?
Fehlende Validierungsnachweise können bei Prüfung durch das Gesundheitsamt zu Beanstandungen, Nachbesserungsauflagen und im Wiederholungsfall zu Betriebsuntersagungen führen. Im zivilrechtlichen Schadensfall greift § 630h BGB: Ohne Dokumentationsnachweis über ordnungsgemäße Sterilisation trägt die Praxis die Beweislast für die Keimfreiheit des eingesetzten Instruments — ein praktisch kaum führbarer Beweis.