



Schneller Service
Kostenlose Rückmeldung innerhalb von 24 Stunden
Erfolg durch Erfahrung
Aus über 15.000 Projekten im Jahr wissen wir, worauf es ankommt
Der digitale Marktführer
Unsere Kunden sprechen für uns:
4,9 von 5 Sternen auf Google








Abstract – Instrumentenaufbereitung: RKI-Empfehlungen für Arztpraxen
- Die Instrumentenaufbereitung in der Arztpraxis ist eine gesetzlich geregelte Betreiberpflicht nach § 8 MPBetreibV (Fassung 20.02.2025): Wer die KRINKO/BfArM-Empfehlung 2012/2024 einhält, gilt als ordnungsgemäß aufbereitend und genießt die Konformitätsvermutung nach § 8 Abs. 2 MPBetreibV.
- Die Risikoklassifizierung nach RKI (unkritisch / semikritisch A+B / kritisch A+B+C) bestimmt den Aufbereitungsaufwand: Kritische Produkte erfordern zwingend Sterilisation, thermolabile kritisch-C-Produkte zusätzlich eine QM-Zertifizierung nach DIN EN ISO 13485; bei Zweifeln gilt die nächsthöhere Risikostufe.
- Der Aufbereitungsprozess folgt einer nicht disponibler Schrittfolge — Vorbereitung, Reinigung, Desinfektion, Prüfung, Pflege, Verpackung, Sterilisation, Kennzeichnung, Freigabe — wobei maschinelle Verfahren im RDG nach DIN EN ISO 15883 manuellen stets vorzuziehen sind und jeder Schritt durch eine Standardarbeitsanweisung belegt sein muss.
- Validierung (IQ/OQ/LQ), jährliche Revalidierung, lückenlose Chargendokumentation mit mindestens 5 Jahren Aufbewahrungsfrist sowie Schulungsnachweise des Aufbereitungspersonals sind Pflichtbestandteile des Qualitätsmanagementsystems — fehlende Nachweise machen die gesamte Aufbereitungsleistung im Prüfungsfall angreifbar.
Inhaltsverzeichnis
Rechtliche Grundlagen: Was § 8 MPBetreibV und die KRINKO-Empfehlung vorschreiben
Die zentrale Rechtsgrundlage für die Aufbereitung von Medizinprodukten in der Arztpraxis ist § 8 Abs. 1 MPBetreibV (Fassung 20.02.2025). Danach ist die Aufbereitung von bestimmungsgemäß keimarm oder steril zur Anwendung kommenden Medizinprodukten mit geeigneten validierten Verfahren durchzuführen. Der Erfolg dieser Verfahren muss nachvollziehbar gewährleistet sein. Die Sicherheit von Patienten, Benutzern und Dritten darf nicht gefährdet werden.
Konformitätsvermutung bei Einhaltung der RKI-Empfehlung
§ 8 Abs. 2 MPBetreibV enthält eine praxisrelevante Konformitätsvermutung: Wer die gemeinsame Empfehlung der Kommission für Krankenhaushygiene und Infektionsprävention (KRINKO) beim Robert Koch-Institut (RKI) und des Bundesinstituts für Arzneimittel und Medizinprodukte (BfArM) beachtet, gilt als ordnungsgemäß aufbereitend. Wer nachweislich nach dieser Empfehlung vorgeht, trägt im Streitfall die Beweislast nicht allein.
Die KRINKO-Empfehlung von 2012 wurde durch eine Ergänzung im Epidemiologischen Bulletin 6/2018 aktualisiert. Diese Ergänzung präzisiert den Validierungsbegriff und verweist auf den neu gefassten § 8 MPBetreibV. Im Dezember 2024 erschien zudem die vollständig überarbeitete Anlage 8 zur Aufbereitung thermolabiler Endoskope. Das RKI führt die Empfehlung daher offiziell als „2012/2024″. Für Praxen ohne Endoskope bleibt das Kerndokument von 2012 inhaltlich maßgeblich.
EU-MDR 2017/745 als übergeordneter Rahmen
Die EU-Medizinprodukteverordnung (MDR 2017/745) bildet den übergeordneten regulatorischen Rahmen. Sie legt grundlegende Sicherheits- und Leistungsanforderungen fest, die auch die Wiederaufbereitung von Instrumenten betreffen (MDR 2017/745, Anhang I, Kapitel III Ziffer 23.1 d). Für den Praxisalltag sind die nationalen Regelwerke — MPBetreibV und KRINKO-Empfehlung — primär handlungsleitend. Die MDR gewinnt vor allem dann an Bedeutung, wenn Einwegprodukte aufbereitet oder externe Aufbereiter beauftragt werden. Für letzteres gilt seit 20.02.2025 der neue § 9 MPBetreibV, der die Aufbereitung von Einmalprodukten erstmals eigenständig regelt.
Haftungsrisiko: Die Delegierung der Aufbereitung an medizinisches Fachpersonal entbindet den Praxisinhaber nicht von der Betreiberverantwortung nach § 8 MPBetreibV. Die Verantwortung für geeignete Verfahren und deren Überwachung verbleibt beim Arzt als Betreiber.
Risikoklassifizierung: In welche Kategorie fällt welches Instrument?
Ausgangspunkt jeder sachgerechten Instrumentenaufbereitung ist die Risikoklassifizierung. Die RKI-Empfehlung 2012/2024 unterscheidet drei Grundkategorien, die sich aus der Art des Patientenkontakts ableiten.
- Unkritische Medizinprodukte kommen ausschließlich mit intakter Haut in Berührung, z. B. EKG-Elektroden. Reinigung und ggf. Desinfektion genügen — eine Sterilisation ist nicht erforderlich.
- Semikritische Medizinprodukte kontaktieren Schleimhaut oder krankhaft veränderte Haut, z. B. Specula. Sie erfordern mindestens eine Desinfektion mit nachgewiesener bakterizider, fungizider und viruzider Wirkung. Semikritisch-B-Produkte stellen erhöhte Anforderungen, etwa durch schwer zugängliche Lumina oder komplexe Oberflächen.
- Kritische Medizinprodukte penetrieren Haut oder Schleimhaut und gelangen in Kontakt mit Blut oder inneren Geweben — z. B. Wundhaken, MIC-Trokare oder ERCP-Katheter. Sie müssen steril zur Anwendung kommen. Kritisch-C-Produkte, die nicht dampfsterilisierbar sind, unterliegen zusätzlich einer externen Qualitätskontrolle durch Zertifizierung nach DIN EN ISO 13485.

Einstufung der Medizinprodukte nach Risikoklasse
| Klasse | Patientenkontakt | Beispiele | Mindestanforderung Aufbereitung |
|---|---|---|---|
| Unkritisch | Intakte Haut | EKG-Elektroden, Blutdruckmanschette | Reinigung, ggf. Desinfektion |
| Semikritisch A | Schleimhaut / veränd. Haut | Spekulum, Zungenspatel | Reinigung + Desinfektion (bakterizid, fungizid, viruzid) |
| Semikritisch B | Schleimhaut, erhöhte Anforderungen | Flexibles Endoskop | Maschinelle R/D bevorzugt; Sonderanlage Nr. 8 |
| Kritisch A | Penetration Haut/Schleimhaut | Wundhaken, chirurg. Instrumente | Reinigung + Desinfektion + Sterilisation (Dampf 134 °C) |
| Kritisch B | Wie A, mit erhöhten Anforderungen | MIC-Trokar | Maschinelle R/D + Sterilisation; Ausbildungsnachweis |
| Kritisch C | Thermolabile, nicht dampfsterilisierbare MP | ERCP-Katheter | Geeignetes Sterilverfahren + QM-Zertifizierung nach DIN EN ISO 13485 |
Bei Zweifeln über die Einstufung gilt: Das Medizinprodukt ist stets der höheren, kritischeren Risikostufe zuzuordnen (RKI-Empfehlung 2012/2024). Diese Zweifelsfallregel schützt vor einer Unterbewertung des tatsächlichen Infektionsrisikos.
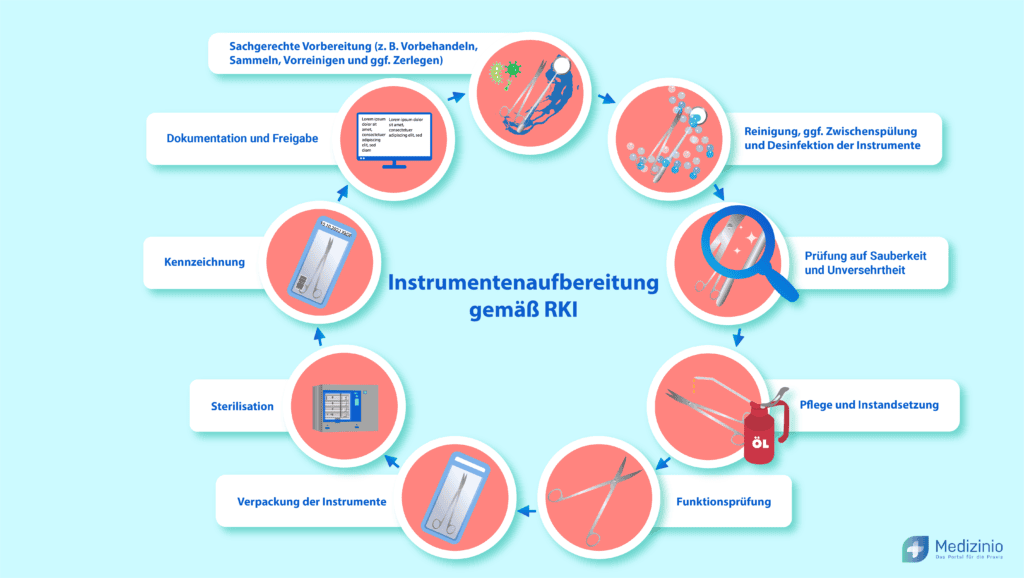
Reihenfolge der Instrumentenaufbereitung: Der Prozessablauf nach RKI
Die Instrumentenaufbereitung ist kein beliebig gestaltbarer Ablauf, sondern ein festgelegter Prozess mit definierter Schrittfolge. Jede Schwäche in einem Einzelschritt gefährdet das Gesamtergebnis — eine unzureichende Reinigung lässt sich durch eine nachfolgende Sterilisation nicht kompensieren (RKI-Empfehlung 2012/2024, Abschnitt 2.2).
Die Reihenfolge gilt für alle angewendeten Medizinprodukte. Nicht jeder Schritt ist für jede Risikoklasse gleichermaßen erforderlich — unkritische Produkte benötigen weder Verpackung noch Sterilisation — die Abfolge selbst bleibt jedoch stets dieselbe.
- Vorbereitung — Grobe Verschmutzungen unmittelbar nach der Anwendung entfernen, Instrumente ggf. zerlegen und sicher verschlossen zum Aufbereitungsort transportieren. Das Antrocknen von Blut und Gewebe ist durch zügige Vorbehandlung zu verhindern.
- Reinigung, Zwischenspülung, Desinfektion, Spülung und Trocknung — Maschinelle Verfahren im RDG sind manuellen vorzuziehen. Eine Zwischenspülung zwischen Reinigung und Desinfektion ist erforderlich, sofern der Hersteller der Prozesschemikalien keine ausreichende Desinfektion ohne Zwischenspülung belegt.
- Prüfung auf Sauberkeit und Unversehrtheit — Sichtkontrolle der Oberflächen, ggf. unter Zuhilfenahme einer Lupenlampe. Bei nicht visuell zugänglichen Flächen ist ein Proteinnachweistest einzusetzen.
- Pflege und Instandsetzung — Instrumente mit Gelenken sind gemäß Herstellerangaben mit geeignetem Pflegemittel zu behandeln.
- Prüfung der technisch-funktionellen Sicherheit — Nachweis, dass die Funktionsfähigkeit des Medizinproduktes durch den Aufbereitungsprozess nicht beeinträchtigt wurde (§ 8 MPBetreibV; RKI-Empfehlung 2012/2024, Anlage 2).
- Verpackung — Nur für Produkte, die steril zur Anwendung kommen. Das Sterilbarrieresystem muss auf das Sterilisationsverfahren und die vorgesehene Lagerung abgestimmt sein.
- Sterilisation — Ausschließlich für kritische Medizinprodukte (A, B, C). Bevorzugtes Standardverfahren: Dampfsterilisation bei 134 °C.
- Kennzeichnung — Chargennummer, Sterilisierdatum, Sterilgutlagerfrist und Freigabeentscheidung müssen aus der Kennzeichnung hervorgehen.
- Dokumentation und Freigabe — Die Aufbereitung endet mit der dokumentierten Freigabe zur Anwendung. Nur eine zur Freigabe schriftlich berechtigte Person darf diese erteilen (RKI-Empfehlung 2012/2024, Abschnitt 2.2.7).
Haftungsrisiko: Die Reihenfolge ist nicht disponibel. Eine Sterilisation ohne vorherige vollständige Reinigung und Desinfektion erfüllt die Anforderungen der RKI-Empfehlung 2012/2024 nicht — und entfaltet keine Konformitätsvermutung nach § 8 Abs. 2 MPBetreibV.
Die Aufbereitungsschritte im Überblick: Was in jeder Praxis lückenlos ablaufen muss
Die Instrumentenaufbereitung folgt einem definierten Prozessablauf. Jeder Schritt ist Bestandteil des validierten Gesamtprozesses. Ein unzulänglich ausgeführter Einzelschritt mindert die Qualität des gesamten Aufbereitungsergebnisses.
Vorbereiten, Sammeln, Vorreinigen, Zerlegen
Unmittelbar nach der Anwendung beginnt die Aufbereitung mit dem sachgerechten Vorbehandeln. Grobe Verschmutzungen werden entfernt, das Instrument wird ggf. zerlegt und in sicher umschlossenen Behältnissen zum Aufbereitungsort transportiert. Dieser Schritt verhindert das Antrocknen von Blut und organischen Rückständen. Angetrocknete Rückstände erschweren die nachfolgende Reinigung erheblich. Für jede dieser Teilhandlungen ist eine Standardarbeitsanweisung zu erstellen (RKI-Empfehlung 2012/2024, Anlage 1).
Persönliche Schutzausrüstung: Was das Personal tragen muss
Während der gesamten Aufbereitung ist Persönliche Schutzausrüstung (PSA) gemäß der Technischen Regel für Biologische Arbeitsstoffe (TRBA 250) Pflicht. Die konkreten Anforderungen richten sich nach dem jeweiligen Arbeitsschritt.

Bei der Eingabe kontaminierter Instrumente in das RDG sowie bei der manuellen Vorreinigung sind flüssigkeitsdichte Schutzkleidung und Einmalhandschuhe erforderlich. Bei vollständig manueller Reinigung und Desinfektion kommen zusätzlich langstulpige Schutzhandschuhe sowie Augen- und Mundschutz hinzu. Das Handschuhmaterial ist auf das eingesetzte Reinigungs- und Desinfektionsmittel abzustimmen. Bei scharfkantigen Instrumenten sind schnittfeste oder schnitthemmende Handschuhe zu verwenden.
Die Aufbereitung von Instrumenten fällt gemäß § 2 Abs. 14 BioStoffV überwiegend in Schutzstufe 2. Bei Instrumenten mit Kontakt zu Tuberkulose-Erkrankten kann das Tragen einer FFP2-Maske erforderlich werden. Bei Patienten mit Creutzfeldt-Jakob-Erkrankung (CJK) gelten die gesonderten Vorkehrungen gemäß Anlage 7 der RKI-Empfehlung 2012/2024.
Reinigung und Desinfektion: maschinell vor manuell
Die RKI-Empfehlung 2012/2024 formuliert eine klare Präferenz: Für kritische und semikritische Medizinprodukte ist die maschinelle Reinigung und Desinfektion im Reinigungs-Desinfektionsgerät (RDG) nach DIN EN ISO 15883 zu bevorzugen. Maschinelle Verfahren bieten eine standardisierte, reproduzierbare und vollständig dokumentierbare Reinigungsleistung.
Die manuelle Instrumentenaufbereitung ist nur zulässig, wenn eine Standardarbeitsanweisung mit nachgewiesener Wirksamkeit vorliegt. Die Leitlinie zur Validierung der manuellen Reinigung und Desinfektion (DGSV/AKI) definiert Installationsqualifikation (IQ), Betriebsqualifikation (OQ) und Leistungsqualifikation (LQ) als Mindestanforderung. Für semikritisch-B- und kritisch-B-Produkte ist die Reinigungsleistung zusätzlich anhand standardisiert mit Blut verschmutzter Prüfkörper (Crile-Klemmen) zu belegen.
Von der Prüfung bis zur Freigabe
Nach Reinigung und Desinfektion folgen: Prüfung auf Sauberkeit und Unversehrtheit — ggf. mit Lupenlampe und semiquantitativem Proteinnachweistest —, Pflege und Instandsetzung sowie Funktionsprüfung. Daran schließen sich Verpackung, Sterilisation, Kennzeichnung und dokumentierte Freigabe an. Erst mit der Freigabe gilt das Instrument als für die Anwendung freigegeben.
Validierungspflicht: Was Praxen wirklich nachweisen müssen
Validierung bedeutet nach § 8 Abs. 1 MPBetreibV: der Nachweis, dass ein Aufbereitungsverfahren ein definiertes Ergebnis — Sauberkeit, Keimarmut oder Sterilität sowie Funktionalität — reproduzierbar und nachweisbar erbringt (06_18.pdf). Ein unzulänglich validierter Einzelschritt gefährdet das Gesamtergebnis.
Installationsqualifikation, Betriebsqualifikation, Leistungsqualifikation
Die Validierung gliedert sich in drei Qualifikationsstufen:
- Installationsqualifikation (IQ): Nachweis, dass Arbeitsbereich, Geräte und Umgebung korrekt installiert und für die Aufbereitung geeignet sind.
- Betriebsqualifikation (OQ): Nachweis, dass Geräte und Prozesse unter definierten Betriebsbedingungen korrekt funktionieren — inkl. Prüfung der Desinfektionsbedingungen, Chemikaliendosierung und Wasserqualität.
- Leistungsqualifikation (LQ): Nachweis der Reinigungswirksamkeit unter praxisüblichen Bedingungen mit betriebstypischer Beladung.
Für Praxen der AEMP-Kategorie A kann der Validierungsumfang durch geeignete Herstellerangaben nach DIN EN ISO 17664 reduziert werden (RKI-Empfehlung 2012/2024, Abschnitt 1.3).
Periodische Revalidierung
Die DIN EN ISO 15883 empfiehlt eine erneute Leistungsqualifikation im Jahresabstand. Eine Revalidierung ist darüber hinaus erforderlich, wenn Routinekontrollen Abweichungen von den Validierungsdaten anzeigen oder wenn Prozess, Gerät oder Beladung wesentlich geändert wurden. Weicht der Betreiber vom Jahresabstand ab, ist dies schriftlich zu begründen. (Quelle: Leitlinie von DGKH, DGSV und AKI für die Validierung und Routineüberwachung maschineller Reinigungs- und thermischer Desinfektionsprozesse für Medizinprodukte)
Tipp: Koordinieren Sie die Revalidierung zeitlich mit der Gerätewartung, um zusätzliche Ausfallzeiten zu vermeiden — die RKI-Empfehlung 2012/2024 empfiehlt diese Vorgehensweise ausdrücklich.
AEMP-Kategorien: Welche Anforderungen gelten für Arztpraxen konkret?
Die RKI-Empfehlung 2012/2024 systematisiert Aufbereitungseinheiten für Medizinprodukte (AEMP) in drei Kategorien. Diese Kategorien folgen direkt aus der Einstufung der aufzubereitenden Medizinprodukte.
AEMP-Kategorien A, B und C im Vergleich
| AEMP-Kategorie | Aufzubereitende MP bis | Typische Einrichtungen | Raumkonzept | Kerntechnik |
|---|---|---|---|---|
| A | Semikritisch A, kritisch A | Allgemein- und Facharztpraxen (ohne OP, ohne Endoskopie) | Eigener Bereich; Zonentrennung unrein/rein/Lagerung (zeitliche Trennung möglich) | RDG und Kleinsterilisator je nach Profil; ggf. Ultraschallbad |
| B | Semikritisch B, kritisch B | Ambulantes Operieren, Endoskopie, Zahnarztpraxen | Eigene Aufbereitungsräume; räumliche Bereichstrennung unrein/rein/Lagerung | RDG(-E), Ultraschallbad, Siegelgerät, validierter Sterilisator |
| C | Kritisch C | Ausgewählte Krankenhäuser; Aufbereiter für Dritte | Separate Räume je Bereich; spezielle technische Anforderungen | Geräte für spezielle Sterilisationsverfahren; Wasseraufbereitungsanlage |
Kategorie A als Regelfall für die niedergelassene Praxis
AEMP-Kategorie A ist der Regelfall für niedergelassene Allgemein- und Fachärzte ohne operative Eingriffe und ohne Endoskopie (RKI-Empfehlung 2012/2024, Anlage 5). Sie setzt einen eigenen Aufbereitungsbereich mit erkennbarer Trennung von unreiner Zone, reiner Zone und Lagerzone voraus. Eine vollständige räumliche Trennung mit separaten Räumen ist für Kategorie A noch nicht zwingend — bei Neu- und Umbauten aber anzustreben.
Praxen mit ambulanten Operationen oder Endoskopie fallen in Kategorie B und erfüllen entsprechend höhere Infrastruktur- und Prozessanforderungen. Die Einstufung ist nicht statisch: Erweitert eine Praxis ihr Leistungsspektrum, muss die AEMP-Kategorie neu bewertet werden.
Externe Aufbereitung: Wann eine Auslagerung sinnvoll ist
Praxen, die die baulichen oder personellen Voraussetzungen für bestimmte Aufbereitungsklassen nicht erfüllen, können die Aufbereitung an einen externen Dienstleister auslagern. Dies ist insbesondere dann relevant, wenn kritisch-B- oder kritisch-C-Produkte aufbereitet werden müssen, die eigene AEMP-Infrastruktur der Kategorie B oder C jedoch nicht vorhanden ist — etwa bei ambulant operierenden Praxen im Aufbau.
Externe Aufbereiter müssen die gleichen regulatorischen Anforderungen erfüllen wie interne Aufbereitungseinheiten. Für Aufbereiter, die für Dritte tätig sind, besteht zudem eine Anzeigepflicht gemäß MPBetreibV. Der Betreiber bleibt auch bei externer Aufbereitung verantwortlich: Die Auswahl eines geeigneten Dienstleisters, die vertragliche Fixierung der Aufbereitungsmodalitäten und die Dokumentation der Übergabe sind Betreiberpflichten, die nicht delegiert werden können (DGSV-Leitlinie Transport/Lagerung; RKI-Empfehlung 2012/2024).
Tipp: Halten Sie bei externer Aufbereitung Übergabenachweis, Transportbedingungen und Rückverfolgbarkeit der Chargen schriftlich fest — diese Unterlagen sind im Prüfungsfall ebenso vorzulegen wie die interne Chargendokumentation.
Verpackung, Lagerung und Transport: Sterilität bis zur Anwendung sicherstellen
Ein aufbereitetes Instrument verliert seinen Sterilstatus, sobald die Verpackungsintegrität kompromittiert wird. Verpackung, Lagerung und Transport sind daher Teil des validierten Aufbereitungsprozesses — nicht nachgelagerte Logistikfragen.
Sterilbarrieresystem: Anforderungen an Verpackung und Siegelnaht
Das Verpackungssystem ist auf das gewählte Sterilisationsverfahren, die Eigenschaften des Instruments sowie die vorgesehene Lagerung und den Transport abzustimmen (RKI-Empfehlung 2012/2024, Abschnitt 2.2.4). Die Dichtigkeit der Siegelnähte ist mindestens durch Sichtprüfung im Verlauf der Aufbereitung zu belegen. Ergänzend gelten die Anforderungen der Normenreihe DIN 58953.
Sterilbarriere- und Verpackungssysteme dürfen nicht feucht gereinigt oder desinfiziert werden. Beim Umgang mit verpacktem Sterilgut sind desinfizierte und trockene Hände erforderlich (DGSV-Leitlinie Transport/Lagerung).
Lagerfristfestlegung und First-in-first-out
Eine einheitliche gesetzliche Lagerfrist existiert nicht. Der Betreiber legt die Lagerfrist eigenverantwortlich fest — anhand einer Risikobewertung der Lagerbedingungen und des eingesetzten Verpackungssystems (DGSV-Leitlinie Transport/Lagerung, Abschnitt 6.3; DIN 58953-8, Kapitel 7.2). Bei geeigneter Verpackungsqualität und sachgerechten Lagerbedingungen sind Fristen von mehr als sechs Monaten dokumentierbar (RKI-Empfehlung 2012/2024).
Die Lagerhaltung folgt dem Prinzip „first in – first out“. Regelmäßige Kontrollen der Verfallsdaten — z. B. monatlich oder vierteljährlich — sind festzulegen, durchzuführen und zu dokumentieren. Instrumente mit abgelaufener Lagerfrist werden einer erneuten Instrumentenaufbereitung zugeführt, bevor sie angewendet werden.
Sterilgut ist staubgeschützt, sauber, trocken, frei von Ungeziefer, bei Raumtemperatur und vor direkter Sonneneinstrahlung zu lagern — keine Lagerung auf Fensterbänken (DGSV-Leitlinie Transport/Lagerung, Abschnitt 5.5).
Dokumentation und Qualitätsmanagement: Was im Prüfungsfall vorgelegt werden muss
Ohne lückenlose Dokumentation ist die Instrumentenaufbereitung rechtlich nicht belegbar. § 8 MPBetreibV und die RKI-Empfehlung 2012/2024 fordern ein Qualitätsmanagementsystem mit Standardarbeitsanweisungen, Chargendokumentation und Schulungsnachweisen.
Standardarbeitsanweisungen für jeden Aufbereitungsschritt
Für jeden Prozessschritt — von der Vorbehandlung über Reinigung, Desinfektion, Verpackung und Sterilisation bis zur Freigabe — ist eine Standardarbeitsanweisung (SAW) zu erstellen und aktuell zu halten (RKI-Empfehlung 2012/2024, Anlage 1). Die SAW legt Zielgröße, Verfahren, verantwortliches Personal und Prüfintervall fest. Sie ist die Grundlage des validierten Prozesses. Eine nicht eingehaltene SAW invalidiert de facto das Validierungsergebnis.
Mindestdokumentation für eine Kategorie-A-Praxis
- Risikoklassifizierung aller aufbereiteten Instrumente (mit Einstufungsgrundlage)
- Standardarbeitsanweisungen für alle Aufbereitungsschritte
- Validierungsprotokoll (IQ, OQ, LQ) für RDG und Sterilisator
- Chargendokumentation je Sterilisationslauf (Chargennummer, Datum, Gerät, Prozessparameter, Freigabe)
- Dokumentation chargenbezogener Routineprüfungen (z. B. Bowie-Dick-Test, Vakuumtest)
- Nachweis über regelmäßige Wartung und Kalibrierung der eingesetzten Geräte
- Schulungs- und Einweisungsnachweise für das mit der Aufbereitung betraute Personal (MPBetreibV)
- Lagerfristfestlegung mit Risikobewertungsgrundlage
- Protokolle der Verfallsdatum-Kontrollen
Die Aufzeichnungen über die Aufbereitung von Medizinprodukten sind nach § 8 MPBetreibV in Verbindung mit der RKI-Empfehlung 2012/2024 mindestens 5 Jahre aufzubewahren. Die Unterlagen sind den zuständigen Behörden auf Verlangen vorzulegen. Nachträgliche Änderungen an Eintragungen sind nur zulässig, wenn erkennbar bleibt, ob sie während oder nach der ursprünglichen Erfassung vorgenommen wurden. Die Aufzeichnungen können auch auf Bild- oder Datenträgern geführt werden, sofern Verfügbarkeit und Lesbarkeit während der gesamten Aufbewahrungsfrist sichergestellt sind (RKI-Empfehlung 2012/2024, Abschnitt 2.2.8).
Haftungsrisiko: Bei kritisch-B-Produkten fordert die RKI-Empfehlung 2012/2024 zusätzlich den Nachweis einer anerkannten Ausbildung des mit der Aufbereitung betrauten Personals (Anlage 6 Sachkenntnis). Fehlt dieser Nachweis, ist die Aufbereitungsleistung für diese Produktklasse angreifbar.
FAQ: Häufige Fragen zur Aufbereitung von Instrumenten in der Praxis
Darf ich kritische Instrumente in der Praxis manuell reinigen und sterilisieren?
Die manuelle Reinigung und Desinfektion kritischer Instrumente ist zulässig, wenn sie nach einer dokumentierten Standardarbeitsanweisung mit nachgewiesener Wirksamkeit durchgeführt wird. Für kritisch-A-Produkte ist die maschinelle Reinigung und Desinfektion bevorzugt. Kritisch-B-Produkte verlangen grundsätzlich maschinelle Reinigung und thermische Desinfektion im RDG. Manuelle Verfahren erfüllen die Anforderungen an Standardisierung und Validierbarkeit für diese Klasse nur unzulänglich (RKI-Empfehlung 2012/2024).
Wie lange darf aufbereitetes Sterilgut in der Praxis gelagert werden?
Eine einheitliche gesetzliche Lagerfrist existiert nicht. Der Betreiber legt sie eigenverantwortlich fest — anhand einer Risikobewertung der Lagerbedingungen und des Verpackungssystems (DGSV-Leitlinie Transport/Lagerung, Abschnitt 6.3; DIN 58953-8, Kapitel 7.2). Bei geeigneter Verpackungsqualität sind Fristen von mehr als sechs Monaten dokumentierbar. Die festgelegte Frist ist schriftlich zu begründen und regelmäßig zu überprüfen.
Was gilt, wenn der Hersteller keine Aufbereitungsangaben nach DIN EN ISO 17664 liefert?
Die DIN EN ISO 17664 verpflichtet Hersteller, ausreichende Informationen für die sachgerechte Instrumentenaufbereitung bereitzustellen. Fehlen diese Angaben, ist der Hersteller zur Vervollständigung aufzufordern. Bei begründetem Verdacht auf ein Vorkommnis ist eine Meldung an das BfArM nach §§ 76 ff. MPDG zu prüfen. Bis zur Klärung darf das Instrument nur aufbereitet werden, wenn ein geeignetes Verfahren auf anderem Wege belegt werden kann (RKI-Empfehlung 2012/2024, Abschnitt 1.2.2).
Was ist der Unterschied zwischen Desinfektion und Sterilisation?
Beide Verfahren reduzieren die Keimbelastung auf einem Instrument — sie verfolgen jedoch unterschiedliche Ziele und erzielen unterschiedliche Keimreduktionsgrade. Wer die gesamte Aufbereitungskette verstehen will, findet im Überblick zu Reinigung, Desinfektion und Sterilisation alle drei Prozessschritte normkonform aufbereitet.
Bei der Desinfektion wird die Keimzahl so weit reduziert, dass von dem Instrument kein Infektionsrisiko mehr ausgeht. Das Ziel ist Keimarmut, nicht Keimfreiheit. Als Maß gilt eine Reduktion um mindestens 5 Log₁₀-Stufen (10⁻⁵). Es wird zwischen chemischer, thermischer und chemothermischer Desinfektion unterschieden. Thermische Verfahren laufen im RDG ab; chemothermische Verfahren kombinieren Desinfektionsmittel mit feuchter Hitze und kommen insbesondere bei thermolabilen Produkten wie flexiblen Endoskopen zum Einsatz.
Die Sterilisation geht darüber hinaus: Alle Mikroorganismen in jedem Entwicklungsstadium einschließlich Sporen werden irreversibel abgetötet. Ein Instrument gilt gemäß DIN EN 556 als steril, wenn die Wahrscheinlichkeit eines vermehrungsfähigen Keims kleiner als 1:1.000.000 ist (≥ 6 Log₁₀-Stufen, 10⁻⁶). Die Dampfsterilisation im B-Autoklav bei 134 °C ist das von der RKI-Empfehlung 2012/2024 bevorzugte Standardverfahren, da es gegenüber potenziellen Einflussfaktoren am robustesten ist.
Sterilisation ist ausschließlich für Medizinprodukte der Klassen kritisch A, B und C vorgeschrieben. Semikritische Produkte erfordern Desinfektion; unkritische Produkte genügen mit Reinigung und ggf. Desinfektion.