




Schneller Service
Kostenlose Rückmeldung innerhalb von 24 Stunden
Erfolg durch Erfahrung
Aus über 15.000 Projekten im Jahr wissen wir, worauf es ankommt
Der digitale Marktführer
Unsere Kunden sprechen für uns:
4,9 von 5 Sternen auf Google





Abstract – Semikritische Medizinprodukte: Klassifikation & Aufbereitungspflichten
- Mit der MPBetreibV-Neufassung (Februar 2025) und der aktualisierten KRINKO-Anlage 8 gelten verschärfte Anforderungen für die Aufbereitung semikritischer Produkte; Fehleinstufungen können Bußgelder bis 30.000 € sowie zivilrechtliche Haftungsfolgen auslösen.
- Semikritische Produkte werden nach dem Spaulding-System in Gruppe A (einfacher Schleimhautkontakt, z. B. Vaginalspekulen, Laryngoskopspatel) und Gruppe B (komplexe Hohlraumstrukturen, z. B. flexible Endoskope) unterteilt – beide Gruppen stellen unterschiedliche Verfahrensanforderungen: Gruppe A erfordert validierte Desinfektion (voll viruzid), Gruppe B zwingend maschinelle Aufbereitung im validierten RDG-E.
- Die Validierungspflicht nach § 8 MPBetreibV umfasst schriftliche Standardarbeitsanweisungen (SAA), regelmäßige Proteinrückstandstests und – bei vernetzten Geräten – explizite IT-Sicherheitsprüfungen; mindestens 50 % des aufbereitenden Personals müssen eine fachspezifische Zusatzqualifikation mit alle zwei Jahre gesetzlich geforderter Auffrischung nachweisen.
- Bei Einstufungszweifeln ist nach KRINKO/BfArM stets die höhere Risikostufe zu wählen; die Entscheidung samt Begründung muss als Teil des Qualitätsmanagementsystems schriftlich dokumentiert werden.
Inhaltsverzeichnis
Was sind semikritische Medizinprodukte — Definition nach RKI
Semikritische Produkte werden als solche definiert, die bestimmungsgemäß mit Schleimhaut oder krankhaft veränderter Haut in Berührung kommen. Im Zuge der Harmonisierung mit der MDR EU 2017/745 (Anhang XVI) umfasst dieser Begriff nun auch Produkte ohne medizinische Zweckbestimmung, wie etwa Instrumente der ästhetischen Chirurgie.
Die Abgrenzung ist strikt:
- Unkritisch: Kontakt nur mit intakter Haut.
- Semikritisch: Kontakt mit Schleimhaut/krankhaft veränderter Haut.
- Kritisch: Durchdringen von Haut/Schleimhaut oder Kontakt mit Blut bzw. inneren Geweben.
Rechtliche Verankerung und Terminologie
Die Einstufungspflicht basiert auf § 8 der MPBetreibV. Wichtig ist die neue Terminologie: Der Begriff „Medizinprodukt“ wurde weitgehend durch „Produkt“ ersetzt; der behandelnde Akteur wird nun rechtlich als „Benutzer“ bezeichnet.
Der Betreiber ist verpflichtet, für jedes Produkt die Art der Aufbereitung schriftlich festzulegen. Hierbei müssen die Herstellerangaben nach DIN EN ISO 17664 sowie der aktuelle Stand der Technik (KRINKO 2024/2026) zwingend einbezogen werden.
Das Drei-Kategorien-Modell: unkritisch, semikritisch, kritisch
Das RKI-Klassifikationsmodell basiert auf dem international etablierten Spaulding-Klassifikationssystem aus den 1960er-Jahren, das den Zusammenhang zwischen Kontaktzone und erforderlichem Desinfektions- bzw. Sterilisationsniveau beschreibt. Die deutsche Adaptation durch KRINKO und BfArM hat dieses Modell auf die Anforderungen der modernen Aufbereitungspraxis übertragen.
| Risikogruppe | Kontaktzone | Beispiele | Aufbereitungsziel |
| Unkritisch | Intakte Haut | EKG-Elektroden, Manschetten | Sauberkeit/Keimarmut |
| Semikritisch A | Schleimhaut | Spekula, Mundspiegel | Validierte Desinfektion (voll viruzid) |
| Semikritisch B | Schleimhaut (komplex) | Flexible Endoskope | Zwingend maschinell (RDG-E) |
| Kritisch A | Wunden/Blutbahn | Skalpelle, Wundhaken | Sterilisation (Dampf) |
| Kritisch B | Wunden (komplex) | MIC-Instrumente | Maschinelle Reinigung + Sterilisation |
Der historische Ursprung: Spaulding-Klassifikation
Dr. Earl H. Spaulding entwickelte das Drei-Stufen-Modell 1968 ursprünglich für die Klassifikation von Patientenpflegeartikeln in Hinblick auf das erforderliche Dekontaminationsniveau. Das Modell unterschied „non-critical“, „semi-critical“ und „critical items“ nach dem Infektionsrisiko durch den jeweiligen Körperkontakt. Die KRINKO übernahm diese Logik und adaptierte sie auf die deutschen Rechtsvorgaben (MPG, MPBetreibV) und die aktuelle Verfahrenstechnik.
Semikritisch A vs. semikritisch B: Die Unterschiede im Detail
Die Binnendifferenzierung der semikritischen Kategorie in Gruppe A und Gruppe B ist für die tägliche Aufbereitungspraxis entscheidend, da beide Gruppen unterschiedliche Verfahrensanforderungen auslösen.
Semikritisch A — Kontakt ohne besondere Anforderungen
Produkte der Kategorie Semikritisch A zeichnen sich dadurch aus, dass sie keine besonderen Anforderungen an die Aufbereitung stellen, da sie beispielsweise keine schwer zugänglichen Lumen oder Hohlräume aufweisen. Die Aufbereitung umfasst Reinigung und Desinfektion, wobei das Wirkspektrum zwingend bakterizid (inklusive Mykobakterien), fungizid und viruzid sein muss. Hier werden Unterschiede zu Reinigung, Desinfektion, Sterilisation und deren Unterschiede erklärt.
Die Analyse der aktuellen Rechtsprechung und der behördlichen Anforderungen zeigt, dass die Validierung dieser Prozesse — auch bei manueller Aufbereitung — mittlerweile unumgänglich ist. Während früher oft eine einfache Dokumentation der Einwirkzeiten als ausreichend erachtet wurde, fordern Gesundheitsbehörden heute den Nachweis der Prozessstabilität durch validierte Standardarbeitsanweisungen (SAA) und regelmäßige Proteinrückstandstests im Rahmen der Leistungsqualifikation.
Typische Produkte der Gruppe A sind Vaginalspekulen, Laryngoskopspatel, starres Rektoskop und intraoral eingesetzte Instrumente ohne Schleimhautpenetration.
Semikritisch B — Kontakt mit erhöhten Anforderungen
Die Kategorie Semikritisch B umfasst Produkte mit erhöhten Anforderungen, insbesondere solche mit Hohlräumen oder komplexen Oberflächenstrukturen, bei denen die Reinigungsleistung nicht durch bloße Inspektion beurteilt werden kann. Für diese Gruppe ist die maschinelle Reinigung und Desinfektion in einem validierten Reinigungs- und Desinfektionsgerät (RDG) mittlerweile als verbindlicher Stand der Technik anzusehen.
Ein besonderes Augenmerk gilt den flexiblen Endoskopen, die als Prototyp der Gruppe Semikritisch B fungieren. Die KRINKO hat in der Neufassung der Anlage 8 (2024) klargestellt, dass die Aufbereitung dieser Geräte nur in speziell dafür ausgestatteten Einheiten (AEMP für Endoskope, früher ZSVA) erfolgen darf, die über die notwendige technische Infrastruktur zur Validierung der Kanalreinigung verfügen.
Tipp: Bei Zweifeln über die Zuordnung zu Gruppe A oder B ist nach der KRINKO/BFARM-Empfehlung 2012/2024 stets die höhere Risikostufe zu wählen. Dokumentieren Sie die Einstufungsentscheidung und Ihre Begründung schriftlich als Teil des Qualitätsmanagementsystems.
Welche Medizinprodukte fallen konkret in die Kategorie semikritisch
Die Zuordnung eines Produkts zur semikritischen Kategorie hängt primär von der vorgesehenen Anwendung ab. Neu ist nach MPBetreibV 2025, dass auch Produkte ohne medizinische Zweckbestimmung (gemäß Anhang XVI MDR), wie Geräte zur Fettabsaugung oder ästhetische Instrumente, in diese Klassifizierung fallen, sofern sie Kontakt mit Schleimhaut haben.
Gruppe A: Typische Praxisprodukte
- Vaginalspekulen und Laryngoskopspatel: Kontakt mit Schleimhäuten ohne Hohlräume.
- Starre Rektoskope: Bei Anwendung auf Schleimhaut ohne Penetration.
- Mundspiegel und Sonden: In der zahnärztlichen Grundversorgung (ohne Schleimhautpenetration).
- Spirometriemundstücke: Kontakt mit der Schleimhaut der Atemwege.
- Besonderheit Ultraschallsonden: Transvaginale oder endokavitäre Sonden werden in der neuen Anlage 8 (2024) differenzierter im „Informativen Anhang 2“ geführt. Hier ist besonders auf Materialverträglichkeit bei automatisierten Verfahren (z. B. UV-C oder H₂O₂) zu achten.
Gruppe B: Produkte mit erhöhten Aufbereitungsanforderungen
- Flexible Endoskope: Gastroskope, Koloskope und Bronchoskope.
- Flexible Zystoskope: Nun vollständig in die Anlage 8 integriert.
- Duodenoskope: Erfordern höchste Aufmerksamkeit wegen der komplexen Albarran-Elevator-Mechanismen.
Abgrenzungsprobleme: Wann wechselt ein Produkt in Kategorie kritisch
Der Übergang wird durch die Art der Anwendung bestimmt. Sobald ein Instrument (z. B. ein Bronchoskop) in Kontakt mit Blut, Wunden oder sterilen Körperbereichen (bzw. sterilen Sekretanteilen) kommt, wechselt es in die Kategorie Kritisch.
Sonderrisiko MRE und Prionen: Die Neufassung der Anlage 8 reagiert massiv auf Infektionsausbrüche mit Carbapenemase-produzierenden Enterobakterien (CPE). Bei Patienten mit Verdacht auf Creutzfeldt-Jakob (CJK) müssen validierte, vorzugsweise alkalische Reinigungsverfahren genutzt werden. Fixierende Desinfektionsmittel (z. B. Glutardialdehyd) sind strikt verboten, da sie Prionen auf Oberflächen stabilisieren können.
Aufbereitungsanforderungen für semikritische Medizinprodukte
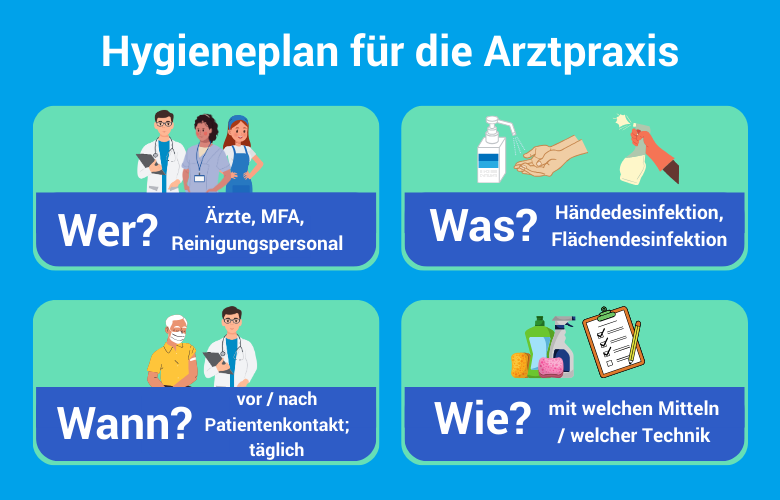
Die Instrumentenaufbereitung umfasst für alle Produktkategorien folgende Kernschritte: Vorbehandeln/Vorreinigen, Reinigung und Desinfektion, Prüfung auf Sauberkeit und Unversehrtheit, Pflege/Instandsetzung, Funktionsprüfung sowie — je nach Erfordernis — Kennzeichnung, Verpackung und Sterilisation. Die Aufbereitung endet mit der dokumentierten Freigabe zur Anwendung.
Anforderungen Gruppe A
Auch für einfache A-Produkte ist die Validierung mittlerweile unumgänglich.
- Wirkungsspektrum: Zwingend bakterizid (inkl. Mykobakterien), fungizid und voll viruzid.
- Prozessstabilität: Auch bei manueller Aufbereitung müssen validierte Standardarbeitsanweisungen (SAA) und regelmäßige Proteinrückstandstests vorliegen.
Anforderungen Gruppe B
Für Produkte der Gruppe B ist die maschinelle Reinigung und Desinfektion in einem validierten RDG-E mittlerweile als verbindlicher Stand der Technik anzusehen.
- Spezialisierung: Die Aufbereitung darf nur in dafür ausgestatteten Einheiten (AEMP für Endoskope) mit technischer Infrastruktur zur Kanalvalidierung erfolgen.
- Trocknung und Lagerung: Gemäß DIN EN 16442 sind für komplexe B-Produkte Lagerungsschränke mit aktiver Belüftung der Kanäle erforderlich, um Rekontaminationen durch Restfeuchte zu verhindern.
Validierungspflicht und Dokumentation
Die Validierung der eingesetzten Aufbereitungsverfahren ist nach MPBetreibV § 8 2025 für semikritische Produkte vorgeschrieben, soweit es sich um Mehrwegprodukte handelt. Für Reinigungs-Desinfektionsgeräte (RDG) gilt DIN EN ISO 15883 als Validierungsgrundlage. Die Ergebnisse der Einstufung und der Risikobewertung des Medizinprodukts sind zu dokumentieren.
- Mikrobiologische Kontrolle: Einführung einer Eingriffsgrenze von 100 KBE pro Endoskop. Bei Überschreitung erfolgt die sofortige Stilllegung des Geräts.
- Personalqualifikation: Gemäß Anlage 8 (2024) müssen mindestens 50 % des Personals eine fachspezifische Zusatzqualifikation vorweisen. Eine Wissensauffrischung ist alle 2 Jahre gesetzlich gefordert.
- IT-Sicherheit: Vernetzte RDG-E unterliegen expliziten IT-Sicherheitsüberprüfungen.
- Software-Updates: Nach Updates mit Funktionsänderungen ist eine erneute Benutzereinweisung gemäß § 17 MPBetreibV Pflicht.
FAQ: Häufige Fragen zur Klassifikation semikritischer Medizinprodukte
Wer legt fest, in welche Kategorie ein Medizinprodukt einzustufen ist?
Die Einstufung obliegt dem Betreiber — also dem Arzt als Praxisinhaber. Dieser hat unter Berücksichtigung der Herstellerangaben (MPBetreibV; DIN EN ISO 17664) schriftlich festzulegen, ob und wie ein Medizinprodukt aufzubereiten ist. Die KRINKO/BfArM-Empfehlung liefert dafür den normativen Orientierungsrahmen. Hersteller können durch ihre Aufbereitungsangaben die Einstufung vorgeben — diese Angaben sind verbindlich zu berücksichtigen.
Gilt die RKI-Klassifikation auch für Einmalartikel?
Einmalartikel dürfen nach § 4 MPBetreibV grundsätzlich nicht aufbereitet werden, sofern der Hersteller dies nicht ausdrücklich vorgesehen hat oder eine Ausnahmeregelung greift. Die Klassifikation nach unkritisch/semikritisch/kritisch ist dennoch relevant, weil sie die Entscheidungsgrundlage bildet, ob eine Wiederaufbereitung überhaupt rechtlich zulässig und technisch durchführbar ist. Für als „Einmal“ gekennzeichnete semikritische Produkte gilt: keine Aufbereitung ohne validierten Nachweis der Prozesseignung und Sicherheit.
Was droht bei fehlerhafter Einstufung eines Medizinprodukts?
Eine Fehleinstufung — z. B. Behandlung eines semikritisch-B-Produktes nach den Anforderungen der Gruppe A — kann zivilrechtliche Haftungsfolgen bei Patientenschäden (§ 280 BGB, Behandlungsvertrag) sowie ordnungswidrigkeitsrechtliche Konsequenzen nach MPG/MPDG auslösen. Darüber hinaus stellt eine unzureichende Aufbereitung von Medizinprodukten einen Qualitätsmangel dar, der im Rahmen von Hygieneprüfungen durch Gesundheitsämter oder bei der KV-Abrechnung relevant werden kann. Ein korrektes Hygienemanagement und die Dokumentationspflicht nach MPBetreibV schützen nur dann, wenn die zugrundeliegende Einstufung korrekt ist.